
Abstract
Better understanding of ependymoma (EPN) biology at relapse is needed to improve therapy at this critical event. Convincing data exist defining transcriptionally distinct posterior fossa (PF) sub-groups A and B at diagnosis. The clinical and biological consequence of these sub-groups at recurrence has not yet been defined. Genome and transcriptome microarray profiles and clinical variables of matched primary and first recurrent PF EPN pairs were used to identify biologically distinct patterns of progression between EPN sub-groups at recurrence. Key findings were validated by histology and immune function assays. Transcriptomic profiles were partially conserved at recurrence. However, 4 of 14 paired samples changed sub-groups at recurrence, and significant sub-group-specific transcriptomic changes between primary and recurrent tumors were identified, which were predominantly immune-related. Further examination revealed that Group A primary tumors harbor an immune gene signature and cellular functionality consistent with an immunosuppressive phenotype associated with tissue remodeling and wound healing. Conversely, Group B tumors develop an adaptive, antigen-specific immune response signature and increased T-cell infiltration at recurrence. Clinical distinctions between sub-groups become more apparent after first recurrence. Group A tumors were more often sub-totally resected and had a significantly shorter time to subsequent progression and worse overall survival. Minimal tumor-specific genomic changes were observed for either PF Groups A or B at recurrence. Molecular sub-groups of PF EPN convey distinct immunobiologic signatures at diagnosis and recurrence, providing potential biologic rationale to their disparate clinical outcomes. Immunotherapeutic approaches may be warranted, particularly in Group A PF EPN.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ependymoma (EPN) is the third most common central nervous system (CNS) tumor in children, accounting for 8–10 % of newly diagnosed pediatric brain tumors. EPN may occur anywhere along the neuroaxis, but is most commonly infratentorial in children. Standard therapy at diagnosis includes maximally safe surgical resection and local radiation therapy, after which most children with EPN will appear to be disease-free. However, nearly half of newly diagnosed children recur [15], and survival after recurrence is poor [16].
Many clinical and molecular prognostic factors for pediatric EPN have been proposed, though none, with the exception of “second look surgery” to achieve gross total resection (GTR) [12], have proven reliable enough to change prognostic stratification or therapeutic management. Success of targeted therapeutic agents has also been limited, in part due to the focus of early phase clinical trials on molecular aberrations in primary EPN with little understanding of the biology of relapsed disease. Better understanding of the biology of EPN at relapse is needed to improve therapy at this critical event.
Convincing data exist defining transcriptionally distinct molecular sub-groups of primary EPN, including two in the posterior fossa (PF) [37, 39]. Phenotypically, Group A tumors occur in younger children and are clinically more aggressive, overexpressing a variety of angiogenesis, inflammatory, and cell proliferation pathways. Group A designation confers increased recurrence risk and poorer overall outcome [39]. Group B tumors are clinically more docile, generally occur in adolescents and young adults, and overexpress genes involved in ciliogenesis, microtubule assembly, and oxidative metabolism [39]. Peyre et al. [30] reported over-expression of genes involved in the kinetochore and neural development and down-regulation of metallothionein genes at recurrence amongst primary and relapsed matched pairs from supratentorial (ST) and infratentorial locations. However, we are the first to investigate clinical and biologic changes in matched diagnostic and recurrent samples in the context of the recently reported PF molecular sub-groups.
We used transcriptome microarray and single nucleotide polymorphism (SNP) analysis to evaluate genomic and transcriptomic alterations in matched primary and first recurrent PF EPN pairs and identified distinct sub-group-specific immunophenotypic changes at recurrence.
Materials and methods
Patients and tumor samples
The initial study cohort consisted of 58 pediatric intracranial EPN samples obtained at either diagnosis (n = 44) or relapse (n = 14) from 44 patients who presented to Children’s Hospital Colorado between 1997 and 2013. Of the 44 primary samples, 35 were located in the PF and 9 were ST. All samples were snap-frozen at the time of surgery and histologically confirmed by our institutional pathologists as EPN according to current World Health Organization (WHO) criteria [6]. Treatment and outcome data were available for all patients. This study was approved by the Institutional Review Board at The University of Colorado Denver (COMIRB 95-500 and 05-149).
Transcriptome microarray analysis
Transcriptomic microarray profiles of tumor samples were generated using Affymetrix HG-U133 Plus 2 GeneChip microarrays (Affymetrix, Santa Clara, CA) as previously described [8]. RNA was isolated from specimens using RNeasy or DNA/RNA AllPrep kit (Qiagen) according to manufacturer’s instructions. Tumor specimens from which both RNA and DNA were isolated were determined by histology to contain ≥70 % tumor cells and thus had minimal normal tissue contamination. RNA quality was verified using the Nano Assay Protocol for the 2100 Bioanalyzer (Agilent) (RNA integrity number ≥8). RNA was amplified, biotin-labeled, and hybridized to Affymetrix HGU133 Plus 2 GeneChips according to manufacturer’s instructions. Analysis of transcriptomic microarray data was performed using Bioconductor functions written in the R programming language (http://www.bioconductor.org). Microarray data CEL files were background corrected and normalized using the guanine cytosine robust Multiarray Average (gcRMA) algorithm, resulting in log2 expression values [41]. To reduce error associated with multiple testing, a filtered list was created containing the highest expressed probe across all samples for each gene that possessed multiple probe sets. This was further filtered to remove probe sets that were expressed below a threshold level that denoted absence of expression in any sample. These microarray data have been deposited in the National Center for Biotechnology Information Gene Expression Omnibus (GEO) database [9] and are publicly accessible through GEO Series accession number GSE50385 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE50385).
Hierarchical clustering (HC) of differentially expressed genes was used to identify molecular sub-groups of EPN. Spearman correlations were used to calculate distances, as implemented by the Bioconductor hclust function. To confirm robust sample clusters, consensus clustering using non-negative matrix factorization was performed (NMF; http://www.genepattern.broadinstitute.org).
Gene ontology analyses
Gene ontologies were analyzed using DAVID (Database for Annotation, Visualization, and Integrated Discovery: http://david.abcc.ncifcrf.gov) [7]. GOTERMs (Gene Ontology Project Terms) [3] were used to evaluate enrichment, defined as greater association with a certain phenotype than would be expected by chance, amongst gene lists. DAVID is a web-based resource that provides Gene Ontology term enrichment scores for lists of genes that have been identified by the user as significantly associated with a particular phenotype or variable. The output is an enrichment score with associated false discovery rate (FDR) adjusted q values and student’s t test p values for each Gene Ontology term.
SNP microarray analysis
DNA was isolated from tumor specimens using DNA/RNA AllPrep kit (Qiagen) according to manufacturer’s instructions. DNA was qualified by Quant-iT PicoGreen dsDNA assay (Life Technologies; Carlsbad, CA), verified to be of high molecular weight on agarose gel, diluted to 50 ng/μl, and labeled for SNP array analysis (HumanOmni 2.5-Quad BeadChip; Illumina; San Diego, CA). Fluorescent signals were imported into GenomeStudio software (Illumina), where the genotype data were normalized and transformed to Log2 R Ratio (LRR) and B allele frequency (BAF) [29]. In analyses for which tumor sample DNA had a matched germline reference (blood DNA) from the same patient, LRR was shown as Log2(R tumor/R reference). In the case of a tumor sample with no matched reference, LRR was represented as Log2(R tumor/R expected), in which R expected was computed from genotype clusters derived from a set of control DNA samples genotyped on the same platform and processed in the same facility as the tumor samples. Those control DNA samples were isolated from 100 blood samples of normal controls. In LRR, any deviations from zero are evidence for copy number change, whereas BAF refers to a normalized measure of relative signal intensity ratio of each allele (assigned as allele A or B). Deviations from the expected values (0.0, 0.5, and 1.0 representing AA, AB, and BB alleles, respectively) are indicative of chromosomal alterations. As described by others [29], signals generated by paired sample analyses showed reduced variability (smaller standard deviation) than single sample analyses.
Data were visualized using Partek Genomic Suite 6.6 software (Partek Inc., St. Louis, MO). To statistically delineate the regions with copy number alterations (CNA) and their breakpoints, genomic segmentation based on the circular binary segmentation algorithm [27] was conducted with Partek. We applied this algorithm for LRR plots setting the following three parameters: minimum genomic markers, p value threshold, and signal-to-noise ratio. Practically, iterative optimizations of parameters were conducted for each case to achieve or approach the most convincing segmentation data. Genomic position is based on human genome assembly (hg19). These data have been deposited in the National Center for Biotechnology Information GEO database [9] and are publicly accessible through GEO Series accession number GSE50876 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE50876).
Immunohistochemistry
Immunohistochemistry (IHC) was performed on 5-μm formalin-fixed paraffin-embedded (FFPE) sections from six Group A and seven Group B primary and recurrent PF tumor pairs. Slides were deparaffinized and subjected to optimal antigen retrieval protocols. Primary antibody against CD4, CD8, and allograft inhibitory factor-1 (AIF-1), a CNS-specific macrophage marker, were used at the following dilutions: 1:250 AIF-1 (01-1974) from Waco Pure Chemicals; 1:2 pre-diluted CD4 (SP35; cat. no. 104R-18) from Cell Marque (Rocklin, CA); and 1:100 CD8 (C8/144B; cat. no. M7103) from Dako. Sections were counterstained with hematoxylin. Each antibody stained a discrete subpopulation of cells that were distributed throughout the tumor parenchyma. Slides were analyzed with an Olympus BX43 microscope using the 40× objective lens and 10× eyepiece (Olympus, Center Valley, CA). Images were captured using an Olympus DP72 camera and cellSens standard 1.5 imaging software. CD4, CD8 and AIF-1 positive cells were counted within high power fields (HPFs) by a single investigator (L.H.). Average infiltration per sample was determined as the mean number of positively staining cells per 10 and 20 400× HPFs for AIF-1 and CD4/CD8, respectively.
Cytokine release assay
Group A and B primary PF EPN samples were collected from routine surgeries at diagnosis. Tumor samples were disaggregated as described previously [17]. Briefly, resected tumor was finely minced with a razor and further triturated by vigorous pipetting. A single cell suspension was obtained by passing the sample through a 70 μm cell strainer of sufficiently large pore size to permit passage of all immune and tumor cells but not clumped tumor cells (Becton–Dickinson, Franklin Lakes, NJ). Heterogeneous distribution of infiltrating immune cells may exist within the tumor margins, but this cannot be addressed once tissue samples have been disaggregated. Rather, disaggregated samples represent a net proportion of cellular subpopulations within the margins of each particular specimen. The majority of processed tumor samples measured at least 1 cm3 in total volume, providing a significant portion of the total tumor mass. Disaggregated cells were viably frozen in standard freezing media containing 10 % DMSO and stored in liquid nitrogen for subsequent analysis. Different immune cell populations may be more or less susceptible to cell death after freeze/thaw, but comparative analysis of data that is generated from identically processed samples minimizes the effect of this potential deficiency.
Characterization of CD4 and CD8 T-cell functionality in Group A and B primary EPN was generated by analysis of cytokine secretion from tumor infiltrating T-cells. Disaggregated tumor preparations were stained with fluorophore-conjugated antibodies CD45, CD3, and CD4 or CD8. T-cells (n = 104) were isolated using Beckman-Coulter MoFlo XDP-100 flow sorter and stimulated for 48 h with PMA/ionomycin in neurobasal A media supplemented with B27, glutamine, EGF, and FGF (Invitrogen, Carlsbad, CA), after which supernatant was collected. A high sensitivity Milliplex Map kit (Millipore) was used to measure the concentration of 13 common cytokines (GM-CSF, IFNγ, IL-1β, IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12 [p70], IL-13, and TNFα).
Statistical analyses
Descriptive statistics were used to evaluate clinical characteristics. Categorical patient characteristics were evaluated by a two-sided Fisher’s exact or Student’s t test. Ordinal variables were evaluated by the Wilcoxon rank sum test. Kaplan–Meier statistics were used to estimate survival probability over time. OS was calculated from the date of initial diagnosis to death from any cause. PFS was calculated from the date of diagnosis to that of first or second progression. Patients who had not had an event (death or progression) at the time of analysis were censored. For all tests, significance was defined as p value <0.05. Raw input data for DAVID was not corrected for multiple testing; however, Benjamini–Hochberg correction was used to globally correct enrichment p values to control FDR for ontologic analysis. Analyses were performed using Prism (GraphPad), R (Bioconductor), and Excel (Microsoft) statistical software.
Results
Identification of transcriptomically distinct primary EPN sub-groups that correspond to previously defined sub-groups
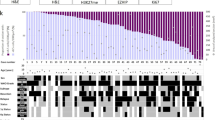
Unbiased HC of the top 5 % of variant genes (883 genes) amongst 44 primary EPN samples confirmed three major transcriptionally distinct sub-groups, including one ST (n = 9) and two PF groups, hereafter referred to as Group A1 (n = 14) and Group B1 (n = 17) (Fig. 1). Four PF samples did not fall into either of the two main PF subgroups and were eliminated from subsequent analyses. The three main sub-groups were confirmed by consensus NMF (Online Resource 1). Concordance between HC and NMF was determined by comparing sub-group location of specific samples within defined groups using each method. Perfect concordance was observed for ST EPN, with all nine samples being grouped together by both HC and NMF. Two PF EPN clusters were generated by NMF, one comprised of thirteen Group A1 by HC and one Group B1 (92.8 % concordance) and the second comprised of thirteen Group B1 and four Group A1 (76.3 % concordance). For subsequent analyses, group designations were based on the results of HC, the same approach adopted by the original studies describing these major PF sub-groups [37, 39].

Transcriptome profiling reveals two posterior fossa EPN sub-groups. Unbiased HC of 44 primary EPN samples based on the top 5 % of variant genes (n = 883) reveals a distinct supratentorial (ST) located tumor sub-group (green) and 2 posterior fossa (PF) subgroups designated Group A1 (red) and B1 (blue). Four samples fell outside of the 3 main clusters and were excluded from further analyses. Below Groups A1 and B1 are a heatmap of NELL2 and LAMA2 expression and color-coded bars to indicate histologic grade (WHO grade II = yellow; WHO grade III = orange), recurrence (recurrent = dark green, non-recurrent = light green), and survival (died of disease = maroon, alive = pink)
Genes that were differentially expressed between the two primary PF sub-groups identified in the present study, Groups A1 and B1, were compared to those described by both Witt et al. [39] and Wani et al. [37]. Ontologies of genes associated with Groups A1 and B1 were identified using DAVID (Table 1). Comparison of sub-group associated ontologies revealed concordance between Group A1 and Group A described by Witt et al. [39] (angiogenesis and other cancer related networks) and Group 1 described by Wani et al. [37] (immune/inflammatory response and regulation of cell proliferation). Group B1 was ontologically similar to Witt et al.’s Group B (ciliogenesis and microtubule assembly). Wani et al. did not find statistically meaningful ontologies that represented Group 2. As added validation of our sub-groups, the genes that best distinguished Groups A and B in Witt et al.’s cohorts and validated in Wani et al.’s cohort were also overexpressed in our respective groups. Specifically, LAMA2 and NELL2 were overexpressed in our Group A1 (2.28-fold; p = 0.007) and Group B1 (11.6-fold; p = 2.30E−05) tumors, respectively. Additionally, Group A1 tumors, when compared to Group B1 tumors, significantly overexpressed all 13 key mesenchymal genes identified in Wani et al.’s Group 1, most notably CHI3L1 (42.0-fold; p = 1.66E−05) and SERPINE1 (23.0-fold; p = 4.00E−07). Since Wani et al. used publically available microarray data from a subset of our tumors to define their initial PF EPN sub-groups, we further validated our sub-groups by confirming concordance of 10 of 12 overlapping samples, including all 5 Group A samples (100 %) and 5 of 7 Group B samples (71 %). This level of concordance is similar to that reported by Wani et al. [37], who compared their microarray-defined sub-groups to those of Witt et al. [39].
Median age at diagnosis was 34 months for Group A1 (range 6–83) and 29 months for Group B1 (range 15–160). Neither age nor gender were significantly different between Groups A and B. Witt et al. and Wani et al. found Group A and Group 1 to be characteristically younger; however, their cohorts included adult as well as pediatric patients, precluding comparison with the present cohort. The proportion of WHO grade II tumors was higher in Group B1, while the opposite was true in Group A1, a difference that showed a trend to significance by contingency analysis (Fisher’s exact p = 0.073). These data are consistent with those of Wani et al., who showed that Group 2, matching the present Group B1, were predominantly WHO grade II, while Group 1 was balanced in respect to grade (Fisher’s exact p = 0.023). The proportion of grade II and III tumors in Groups A and B described by Witt et al. were not different. Other clinical details for primary Groups A1 and B1 PF tumors are included in Table 2.
Evaluation of sub-group-specific genomic changes at diagnosis and recurrence
We used SNP to evaluate CNA amongst 11-matched primary and first recurrent PF EPN pairs (5 Group A, 6 Group B). Similar to Witt et al. [39], who utilized comparative genomic hybridization array, we observed larger and more frequent CNA in Group B1 tumors, whereas Group A1 tumors displayed quieter genomic profiles (Fig. 2a). We observed a larger number of amplifications than deletions in each sub-group at diagnosis and recurrence (Fig. 2b). Interestingly, a larger proportion of our Group B tumors exhibited gain of chromosome 1q (Fig. 2a), which is contradictory to Witt et al. [39] and others [22, 24] who have associated this aberration with poor outcome. This finding likely relates to our small cohort and our selection bias of performing SNP analysis on only Group B tumors that recurred. On the whole, recurrent samples were genomically similar to their matched primary tumors in both Group A (p = 0.14) and Group B (p = 0.71) (Fig. 2b).

Identification of sub-group-specific copy number alterations (CNA) in primary and recurrent posterior fossa EPN genomes. a Genomic analysis using Illumina HumanOmni 2.5-Quad BeadChip SNP microarray reveals fewer CNAs in Group A (top panel) than Group B (bottom panel). Both groups generally conserve their CNAs at recurrence. Sample number is listed on the y-axis (primary = _1, recurrence = _2), and chromosome number is listed on the x-axis. Amplification = red, deletion = blue. b Venn diagrams depict average copy number amplifications and deletions in kilobases. Recurrent Group A (GA2) (n = 5) and Group B (GB2) (n = 6) convey a similar number of CNAs to their primary counterparts, GA1 and GB1, respectively
Characterization of PF Sub-Groups A and B at Recurrence
Recurrent tumors were assigned to Groups A or B based on HC of their matched primary tumors. Recurrent tumors, hereafter referred to as Groups A2 (n = 7) and B2 (n = 7), were compared by transcriptomic analysis to determine whether elements defining Groups A1 and B1 were conserved. To address the question of sub-group “switching” at recurrence, we used a reiterative HC approach to evaluate each recurrence individually in the context of the top 5 % of differentially expressed genes between Groups A1 and B1 (Fig. 1). We observed a change in sub-group designation at recurrence for three of seven Group A tumors (changed to Group B at recurrence) and one of seven Group B tumors (changed to Group A at recurrence). Though our numbers are too small to make conclusions regarding sub-group switching at recurrence and outcome, our data indicate that changing from Group A at diagnosis to Group B at recurrence does not confer a survival advantage, as two of these three patients died of disease (samples 319 and 691) and one has progressive disease (sample 459) (Table 2). The one patient whose sub-group designation changed from Group B at diagnosis to Group A at recurrence (sample 483) also did poorly and ultimately died of disease (Table 2).
Ontologic analysis of genes overexpressed in Group A2 compared to B2 showed minimal overlap with respective ontologies that defined Group A1 in the present study or with the major ontologies distinguishing sub-groups in prior studies [37, 39]. The one exception is ontologic representation of GTPase signaling in both Group A1 tumors (“small GTPase mediated signal transduction”; GOTERM 7264; p = 0.008) and Group A2 tumors (“regulation of RAS GTPase activity”; GOTERM 32318; p = 0.019 and “regulation of GTPase activity”; GOTERM 43087; p = 0.031) (Table 3). Hence, GTPase signaling represents the only “druggable” pathway consistent with Witt et al.’s data in Group A primary tumors [39] that we found conserved at recurrence. Group B2 ontologies did not match those associated with Group B1, apart from “ion transport” (GOTERM 6811; p = 1.91E−04) (Table 4). However, the previously identified marker of Group A tumors, LAMA2, was overexpressed in Group A2 (1.84-fold; p = 0.047), and Group B marker, NELL2, was over-expressed in Group B2 (19.7-fold; p = 0.001). The list of genes characterizing Group A1 (e.g., genes over-expressed ≥2-fold, p < 0.05 in Groups A1 versus B1) was compared in Group A2. Minimal overlap was seen in these gene lists, with only 1.6 % (16 of 1003) of genes distinguishing Group A1 being conserved at recurrence (Fig. 3). Group B demonstrated stronger conservation of signature genes, with 22 % (114 of 633) of Group B1 genes present in the Group B2 signature (Fig. 3). Together, these data demonstrate significant transcriptomic change between presentation and recurrence in both groups, with minor conservation of those elements defining molecular sub-groups at diagnosis.

Genes distinguishing PF EPN sub-groups at recurrence are dissimilar from distinguishing genes at presentation. Venn diagrams depict minimal overlap of genes that define Group A at presentation (up in A1 vs. B1) with genes that define Group A at recurrence (up in A2 vs. B2). Percentages represent proportion of genes in geneset that are overlapped. Group B-defining genes showed stronger overlap by this measure, approximately a quarter of genes being conserved. Immune genes overlapping between Groups A1 and A2 and Groups B1 and B2
Identification of sub-group-specific transcriptomic changes in paired samples between diagnosis and recurrence
Further characterization of the transcriptomic changes between diagnosis and recurrence were performed in a sub-group-specific manner. Transcriptomic characteristics of 14 matched primary and first recurrent EPN pairs (7 Group A, 7 Group B) were analyzed. Differentially expressed genes defined by paired Student’s t test (p < 0.05) and fold change (≥2) between paired primary and recurrent samples were identified and subjected to ontological analysis. The most significant ontological change in Group A tumors was the loss of immune and inflammatory ontologies at recurrence, specifically “response to wounding”, “defense response”, and “inflammatory response” (Table 4). No ontologies associated with genes that increased at recurrence in Group A were significant by FDR. Conversely, Group B1 demonstrated significantly enriched immune-related ontologies in those genes overexpressed at recurrence, notably “immune response” and “response to virus” (Table 4). LAMA2 was decreased in Group A2 tumors (1.91-fold; p = 0.006), while NELL2 did not significantly change in Group B2 tumors.
Divergence of clinical outcome after first recurrence and resectability is associated with sub-group designation at diagnosis
Kaplan–Meier survival analysis was performed on sub-groups derived from unbiased HC at diagnosis. Similar to prior publications [37, 39], 5-year OS was significantly longer in Group B (88 %) than Group A (40 %) (p = 0.02) (Fig. 4a). 5-year PFS from diagnosis to first recurrence was similar between sub-groups in our cohort (Fig. 4b), a finding that differs from Witt et al. [39] and Wani et al. [37], who both reported prolonged time to first progression in Group B tumors. This discrepancy may relate to our small cohort. Interestingly, however, we found significant divergence in PFS between sub-groups following first recurrence, where 5-year PFS was 75 % for Group B and 24 % for Group A (p = 0.02) (Fig. 4c). Potentially correlated with outcome was extent of tumor resection, which is known to hold prognostic significance in EPN [11]. Group A tumors were more likely than Group B tumors to be sub-totally resected at diagnosis (p = 0.08) and recurrence (p = 0.009) (Fig. 4d, e). Since our cohort was derived from a single institution with a common surgical approach, these data suggest unique biologic characteristics of Group A tumors that make them more difficult to completely resect, an effect that appears even more pronounced at recurrence.

Divergence of clinical outcome after first recurrence is associated with PF EPN sub-group designation at diagnosis. Kaplan–Meier plots demonstrate (a) a significantly shorter overall survival (OS) in Group A than Group B PF EPN, (b) no sub-group difference in time to first recurrence after diagnosis, and (c) a significant worse progression-free survival in Group A following first recurrence. Statistical significance was determined by log-rank test. d Resectability of tumors shows no significant sub-group association at diagnosis. e At recurrence, Group A tumors are significantly less resectable
Immunobiology distinguishes sub-groups at diagnosis and evolves in a sub-group-specific fashion at recurrence
In the present study, the predominant ontologies that distinguish Groups A1 from B1 and that change at recurrence within Groups A and B were immune-related (Tables 1, 4). To reiterate, the most significant ontology representing genes overexpressed in Group A1 (vs. Group B1) was “immune response” (GOTERM 6955) (FDR = 1.34E−27) (Table 1). In Group A primary and recurrent matched pairs (n = 7), the most significant ontology representing genes overexpressed in Group A1 (vs. Group A2) that decrease at recurrence (Online Resource 2) was “response to wounding” (GOTERM 9611) (FDR = 7.07E−11). In Group B primary and recurrent matched pairs (n = 7), the most significant ontology representing genes overexpressed in Group B2 (versus Group B1) was “immune response” (GOTERM 6955) (FDR = 1.65E−32) (Table 4). Previous work by our lab implicated the immune system in EPN biology by associating specific immune gene signatures and immune cell tumor infiltration with longer PFS in pediatric EPN [8]. To better elucidate the contribution of immunity in EPN in the current study, more thorough characterization of sub-group and recurrence-specific immunophenotypes was performed utilizing transcriptomic analyses, IHC, and functional immune analyses.
The 1,003 genes overexpressed in Group A1 versus B1 (p < 0.05; fold ≥2) were filtered to identify 125 genes with an “immune response” ontological annotation (GOTERM 6955). Similarly, 30 of the 307 genes that were significantly down-regulated in Group A2 (down in Group A2 vs. A1) and 69 of the 302 genes overexpressed in Group B2 (up in Group B2 vs. B1) were “immune response” annotated. These three “immune response”-annotated gene sets were compared, revealing that the majority of genes overexpressed in Group B2 were discrete from those overexpressed in Group A1 (up in Group A1 vs. B1) and those decreased in Group A2 (down in Group A2 vs. A1) (Fig. 5a). Immune genes that decreased at recurrence in Group A (Online Resource 2) were largely the same “immune response” genes (28 of 30) that distinguished Group A1 from B1 (Fig. 5a) and were predominantly related to inflammation (Fig. 5b).

Posterior fossa EPN reveal distinct sub-group-specific immunophenotypes at diagnosis and recurrence. a Venn diagram of overlapping “immune response” genes that are (i) overexpressed in Group A1 versus B1 (up in A1 vs. B1), (ii) overexpressed in Group B2 versus B1 (up in B2 vs. B1), and (iii) decreased in Group A2 versus A1 (down in A2 vs. A1). B Pie charts representing specific immune-functional categories for genes comprising (i) Group A1 immune signature (n = 93), (ii) Group B2 immune signature (n = 37), and (iii) decreased in Group A2 versus A1 immune genes that overlapped with the Group A1 immune signature (n = 25). c Representative histology of (upper panel) tumor-infiltrating cytotoxic T-cells (CD8; red) and helper T-cells (CD4; brown), and (lower panel) tumor-infiltrating microglia/macrophages (AIF1; brown) in PF EPN (original magnification ×400). d Average tumor-infiltrating CD4 and CD8 positive cells per 20 high power fields (HPFs) and average tumor infiltrating AIF-1 positive cells per 10 HPFs in 6 Group A and 7 Group B matching primary and recurrent pairs. e Significantly higher levels of TNFα, IFNγ and GM-CSF are secreted from Group B1 than Group A1 tumor-infiltrating CD4 helper T-cells
To determine whether genes that distinguished Group B2 from A1 represented a specific immunophenotype, a more detailed analysis of immune-function related genes was performed. Immune signature genes (i.e., immune-related genes exclusive to each group) from Groups A1 (n = 93) and B2 (n = 37) were manually assigned to broad immune functional categories (adaptive, antiviral, immunoregulatory, inflammatory, innate, superoxide generation), if known, based on available published material (Online Resource 3). Those genes associated with major immune functions across multiple categories, or that had minimal known functions, were assigned as “not specific”. This analysis revealed specific immune functional categories that distinguished Group A1 from Group B2 immune gene signatures (Fig. 5b). The Group A1 gene signature was distinguished by a predominance of inflammation-associated (35 %) and immunoregulatory (12 %) genes. Group A1 inflammatory genes included a large number of chemokine, complement system, and interleukin 1–associated genes, and immunoregulatory genes included V-set and immunoglobulin domain containing 4 (VSIG4) and key immunosuppressive gene transforming growth factor beta 1 (TGFβ 1) (Online Resource 3) [36, 40]. Conversely, the Group B2 gene signature was associated with antiviral (30 %) and adaptive (22 %) immune roles. The antiviral signature genes, notably Apolipoprotein B mRNA editing enzyme (APOBEC), oligoadenylate synthetase (OAS) and toll-like receptor 3 (TLR3) were not elevated in Group B1 versus A1, suggesting that there was no preexisting antiviral signature in Group B tumors at diagnosis. The predominant role of these genes is recognition of pathogenic nucleic acids and consequent innate immune responses [1, 26, 34]. The antiviral response in Group B2 may represent innate host cell recognition of cancer-specific aberrant nucleic acid. CD8A and MHC class-II genes represented adaptive immune response (Online Resource 3).
As previously reported by our group, histological analysis of immune function genes identified by transcriptome analysis of EPN whole tumor extracts revealed that their expression was restricted to sub-populations of tumor-infiltrating immune cells [8]. Based on the prominent adaptive immune response signature and overexpression of key T-cell gene CD8A observed in Group B2, it was hypothesized that this sub-group harbored an increased number of infiltrating T-cells. Histological evaluation of immune cell infiltration in Group A and B tumors at diagnosis and recurrence was therefore performed by IHC staining of CD4 and CD8 T-cells and macrophage/microglia in paraffin sections of surgical samples (Fig. 5c). The IHC cohort consisted of 6 Group A and 7 Group B matched primary and recurrent samples. Enumeration of infiltrating CD4 and CD8 T-cells and microglia/macrophages (AIF-1 immunoreactive) demonstrated significantly more abundant CD4 T-cells (2.6-fold) in Group B2 tumors than others (p = 0.009) (Fig. 5d). CD8 T-cells were similarly more abundant in B2 than others (2.4-fold) with a trend toward significance (p = 0.051) (Fig. 5d). Microglia/macrophages were not significantly different in B2 than in other sub-groups.
The predominance of inflammatory and immune-regulatory genes in Group A1, particularly TGFβ1 and VSIG4, suggests that infiltrating immune cells in this sub-group may be functionally deficient or immunosuppressive. To investigate this hypothesis, the functional capacity of CD4 and CD8 T-cells isolated from 12 primary EPN samples (6 Group A and 6 Group B) was evaluated in specific reference to their ability to secrete a panel of 13 common cytokines in response to immune stimulation. Using this approach, CD4 T-cells from Group A1 tumors were shown to secrete significantly lower amounts of key immune-stimulatory cytokines TNFα (2.7-fold), IFN γ (5.3-fold), and GM-CSF (5.1-fold) than Group B1 tumors (Fig. 5e). Interleukin 2 (IL-2) secretion was also 2.8-fold lower in Group A1, which approached significance (p = 0.053). Cytokines IL-1β, IL-4, IL-5, IL-6, IL-7, IL-8, IL-10, IL-12 [p70], and IL-13 were not significantly different between Group A1 and B1. Less IL-2, GM-CSF, TNF, and INFγ was also secreted by CD8 T-cells from Group A1 compared to Group B1, although this only showed a trend toward significance (p = 0.08–0.11). Together, these data suggest impaired immune function in Group A1 tumor infiltrating T-cells.
Discussion
There is accumulating evidence in a variety of human cancers that immunobiology plays an important role in tumor eradication or promotion. To date, the interplay of immunobiologic factors in CNS malignancies has been most extensively studied in adult glioblastoma, which displays inherently suppressed cellular immunity facilitated by tumor-secreted and cell surface immunosuppressive factors and tumor-induced immunosuppressive leukocyte populations [5, 18, 23, 28, 33, 35, 40]. This immunosuppressed phenotype is proposed to dampen the anti-tumor effect conferred by innate host immunity and has been associated with worse clinical outcome [38]. However, there is recent evidence by two groups, both who utilized transcriptome microarray, that immunophenotypic variation exists amongst previously described molecular sub-types of glioblastoma [2, 10]. Immunobiologic characteristics of pediatric brain tumors are less well known.
Despite similar histologic appearance, EPNs often display diverse clinical behavior, which may be due in part to differences in immune phenotype. Good and poor risk sub-groups of EPN were recently defined through genomic and transcriptomic analyses [21, 37, 39]. However, therapeutic translation of these important molecular findings is slow, and still nearly half of children with newly diagnosed EPN relapse [15]. Salvage for some children with relapsed EPN is possible with complete resection and re-irradiation [19], but targeted molecular therapies at relapse have yielded disappointing results [13, 14, 20], highlighting our poor understanding of phenomena that drive recurrence. Recently, our group published an analysis of tumor-infiltrating immune cells in primary pediatric EPN and showed a positive association between macrophage and CD4 infiltration and survival, suggesting that at least some EPNs have a different immunophenotype than that described in adult glioblastoma [8]. The purpose of our study was to determine sub-group designation of recurrent PF EPN, to immunologically characterize sub-groups at diagnosis and recurrence, and to investigate sub-group-specific genomic and transcriptomic changes between diagnostic and recurrent samples to identify potential novel therapeutic approaches to improve survival. Data from our purely pediatric cohort reveal clinically relevant, sub-group-specific immune-related signatures in primary and recurrent PF EPN that support immunotherapeutic approaches as an adjunct to standard therapy in a subset of pediatric EPN.
Genomic comparison of primary and recurrent samples in our cohort revealed relative conservation of CNA in each PF sub-group, suggesting that observed survival differences derive not from large-scale genomic reorganization but from transcriptomic and micro-environmental changes. At a transcriptomic level, we observed general concordance between Groups A1 and B1 and the molecular sub-groups defined by Witt et al. [39] and Wani et al. [37], demonstrated by consistent gene ontologies and overexpression of sub-group defining genes LAMA2 and NELL2 in Groups A1 and B1, respectively. Examination of biological differences between sub-groups at diagnosis suggests that immunophenotype may be of critical importance in the poor outcome of Group A1 tumors. Specific immune genes overexpressed in Group A1, including TGFβ, have a predominantly immunosuppressive phenotype, similar to that of adult glioblastoma. Expression data are supported by examination of the functional capacity of CD4 and CD8 T-cells from Groups A1 and B1, in which CD4, and to a lesser extent CD8, T-cells from Group A1 secreted significantly lower amounts of immune-stimulatory cytokines than Group B1, implying relative immune suppression in Group A1. Our data also strongly suggest the importance of immunophenotype in relapsed EPN, which may account for the stark divergence in outcome observed between PF sub-groups at recurrence. Group B2, in which we observed nearly three times superior PFS than Group A2 after first recurrence (Fig. 4c), dramatically increased their endogenous immune content compared to Group B1. Specifically, Group B2 tumors displayed an activated immune state, noted by overexpression of antigen-specific, immune-stimulatory genes and an increase in tumor-infiltrating CD4 and CD8 T-cells. In contrast, the immunosuppressed phenotype associated with Group A1 appears to preclude development of an activated immune phenotype at recurrence, as observed in Group B2.
Further observations from our study indicate that tumor biology may affect known prognostic factors in EPN, such as extent of tumor resection [4, 32]. We observed a higher incidence of STR in Group A tumors at diagnosis and recurrence (Fig. 4d, e), which we hypothesize relates to overexpression of key mesenchymal genes that are characteristic of a particularly invasive and poorly performing subset of adult glioblastoma [31]. Interestingly, this mesenchymal subset of glioblastoma is partly characterized by overexpression of LAMA2 [25], the representative marker of Group A EPN [37, 39]. Our data are consistent with Wani et al., who reported overexpression of 13 mesenchymal genes in their poor outcome Group 1 tumors [37], and are further supported by Witt et al. [39], who noted ‘invasive growth’ on neuropathologic examination of Group A tumors and observed a survival advantage in Group A tumors that were gross totally resected. In our cohort, both the suppressed immunophenotype and mesenchymal gene expression profile of Group A tumors highlight the influence of tumor microenvironment on their inferior survival.
We are the first to describe sub-group-specific biologic changes in PF EPN at recurrence. Our findings are largely distinct from those of Peyre et al. [30], who identified transcriptomic profiles of progression in EPN from both supra- and infratentorial locations, as they did not distinguish between PF Groups A and B. Comparison of matched primary and first recurrent PF EPN samples enabled identification of unique immunobiologic phenotypes that correlate with outcome at diagnosis and recurrence. Gain of antigen-specific, activated immunity in Group B tumors conferred a survival advantage, suggesting that immunophenotypic reprogramming of Group A tumors from an immune suppressed to an immune activated state may improve outcome. While considerable effort is still needed to characterize immunophenotypic variation in EPN and other pediatric brain tumors, our findings support biologically driven, sub-group-specific immunotherapeutic approaches in primary and recurrent PF EPN.
References
Alexopoulou L, Holt AC, Medzhitov R, Flavell RA (2001) Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 413(6857):732–738
Arimappamagan A, Somasundaram K, Thennarasu K et al (2013) A fourteen gene GBM prognostic signature identifies association of immune response pathway and mesenchymal subtype with high risk group. PLoS One 8(4):e62042
Ashburner M, Ball CA, Blake JA et al (2000) Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat Genet 25(1):25–29
Bouffet E, Perilongo G, Canete A, Massimino M (1998) Intracranial ependymomas in children: a critical review of prognostic factors and a plea for cooperation. Med Pediatr Oncol 30(6):319–29 (discussion 329–31)
Choi C, Xu X, Oh JW, Lee SJ, Gillespie GY, Park H, Jo H, Benveniste EN (2001) Fas-induced expression of chemokines in human glioma cells: involvement of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase. Cancer Res 61(7):3084–3091
David N Louis, Ohgaki H (2007) WHO classification of tumours of the central nervous system [electronic resource]. World Health Organization
Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA (2003) DAVID: database for annotation, visualization, and integrated discovery. Genome Biol 4(5):P3
Donson AM, Birks DK, Barton VN, Wei Q, Kleinschmidt-DeMasters BK, Handler MH, Waziri AE, Wang M, Foreman NK (2009) Immune gene and cell enrichment is associated with a good prognosis in ependymoma. J Immunol 183(11):7428–7440
Edgar R, Domrachev M, Lash AE (2002) Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30(1):207–210
Engler JR, Robinson AE, Smirnov I, Hodgson JG, Berger MS, Gupta N, James CD, Molinaro A, Phillips JJ (2012) Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PLoS One 7(8):e43339
Foreman NK, Love S, Thorne R (1996) Intracranial ependymomas: analysis of prognostic factors in a population-based series. Pediatr Neurosurg 24(3):119–125
Foreman NK, Love S, Gill SS, Coakham HB (1997) Second-look surgery for incompletely resected fourth ventricle ependymomas: technical case report. Neurosurgery 40(4):856–60 (discussion 860)
Fouladi M, Stewart CF, Blaney SM, Onar-Thomas A, Schaiquevich P, Packer RJ, Gajjar A, Kun LE, Boyett JM, Gilbertson RJ (2010) Phase I Trial of Lapatinib in Children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol 28(27):4221–4227
Fouladi M, Stewart CF, Olson J et al (2011) Phase I trial of MK-0752 in children with refractory CNS malignancies: a pediatric brain tumor consortium study. J Clin Oncol 29(26):3529–3534
Gajjar A, Packer RJ, Foreman NK, Cohen K, Haas-Kogan D, Merchant TE, on behalf of the COG Brain Tumor Committee (2012) Children’s Oncology Group’s 2013 blueprint for research: Central nervous system tumors. Pediatr Blood Cancer n/a–n/a
Goldwein JW (1990) Recurrent intracranial ependymomas in children survival, patterns of failure, and prognostic factors. Cancer 66:557–563
Griesinger AM, Birks DK, Donson AM, Amani V, Hoffman LM, Waziri A, Wang M, Handler MH, Foreman NK (2013) Characterization of distinct immunophenotypes across pediatric brain tumor types. J Immunol. doi:10.4049/jimmunol.1301966
Hishii M, Nitta T, Ishida H, Ebato M, Kurosu A, Yagita H, Sato K, Okumura K (1995) Human glioma-derived interleukin-10 inhibits antitumor immune responses in vitro. Neurosurgery 37(6):1160–6 (discussion 1166–7)
Hoffman LM, Reed Plimpton S, Foreman NK, Stence NV, Hankinson TC, Handler MH, Hemenway MS, Vibhakar R, Liu AK (2013) Fractionated stereotactic radiosurgery for recurrent ependymoma in children. J Neurooncol. doi:10.1007/s11060-013-1259-3
Jakacki RI, Hamilton M, Gilbertson RJ, Blaney SM, Tersak J, Krailo MD, Ingle AM, Voss SD, Dancey JE, Adamson PC (2008) Pediatric phase I and pharmacokinetic study of erlotinib followed by the combination of erlotinib and temozolomide: a Children’s Oncology Group Phase I Consortium Study. J Clin Oncol 26(30):4921–4927
Johnson RA, Wright KD, Poppleton H et al (2010) Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature 466(7306):632–636
Kilday JP, Mitra B, Domerg C et al (2012) Copy number gain of 1q25 predicts poor progression-free survival for pediatric intracranial ependymomas and enables patient risk stratification: a prospective European clinical trial cohort analysis on behalf of the children’s cancer leukaemia group (CCLG), societe Francaise d’oncologie pediatrique (SFOP), and international society for pediatric oncology (SIOP). Clin Cancer Res 18(7):2001–2011
Komohara Y, Ohnishi K, Kuratsu J, Takeya M (2008) Possible involvement of the M2 anti-inflammatory macrophage phenotype in growth of human gliomas. J Pathol 216(1):15–24
Korshunov A, Witt H, Hielscher T et al (2010) Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol 28(19):3182–3190
Lathia JD, Li M, Hall PE et al (2012) Laminin alpha 2 enables glioblastoma stem cell growth. Ann Neurol 72(5):766–778
Nilsen TW, Baglioni C (1979) Mechanism for discrimination between viral and host mRNA in interferon-treated cells. Proc Natl Acad Sci USA 76(6):2600–2604
Olshen AB, Venkatraman ES, Lucito R, Wigler M (2004) Circular binary segmentation for the analysis of array-based DNA copy number data. Biostatistics 5(4):557–572
Parsa AT, Waldron JS, Panner A et al (2007) Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med 13(1):84–88
Peiffer DA, Le JM, Steemers FJ et al (2006) High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res 16(9):1136–1148
Peyre M, Commo F, Dantas-Barbosa C et al (2010) Portrait of ependymoma recurrence in children: biomarkers of tumor progression identified by dual-color microarray-based gene expression analysis. PLoS One 5(9):e12932
Phillips HS, Kharbanda S, Chen R et al (2006) Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 9(3):157–173
Robertson PL, Zeltzer PM, Boyett JM et al (1998) Survival and prognostic factors following radiation therapy and chemotherapy for ependymomas in children: a report of the Children’s cancer group. J Neurosurg 88(4):695–703
Rodrigues JC, Gonzalez GC, Zhang L et al (2010) Normal human monocytes exposed to glioma cells acquire myeloid-derived suppressor cell-like properties. Neuro Oncol 12(4):351–365
Sheehy AM, Gaddis NC, Choi JD, Malim MH (2002) Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature 418(6898):646–650
Sippel TR, White J, Nag K, Tsvankin V, Klaassen M, Kleinschmidt-DeMasters BK, Waziri A (2011) Neutrophil degranulation and immunosuppression in patients with GBM: restoration of cellular immune function by targeting arginase I. Clin Cancer Res 17(22):6992–7002
Vogt L, Schmitz N, Kurrer MO et al (2006) VSIG4, a B7 family-related protein, is a negative regulator of T cell activation. J Clin Invest 116(10):2817–2826
Wani K, Armstrong TS, Vera-Bolanos E et al (2012) A prognostic gene expression signature in infratentorial ependymoma. Acta Neuropathol 123(5):727–738
Waziri A (2010) Glioblastoma-derived mechanisms of systemic immunosuppression. Neurosurg Clin N Am 21(1):31–42
Witt H, Mack SC, Ryzhova M et al (2011) Delineation of two clinically and molecularly distinct subgroups of posterior fossa ependymoma. Cancer Cell 20(2):143–157
Wrann M, Bodmer S, de Martin R, Siepl C, Hofer-Warbinek R, Frei K, Hofer E, Fontana A (1987) T cell suppressor factor from human glioblastoma cells is a 12.5-kd protein closely related to transforming growth factor-beta. EMBO J 6(6):1633–1636
Wu Z, Irizarry RA, Gentleman R, Murillo FM, Spencer F (2004) A model based background adjustment for oligonucleotide expression arrays
Acknowledgments
This work was supported by the National Institutes of Health (R01 CA140614), Ruth L. Kirschstein National Research Service Award (T32 CA082086-13), the Tanner Seebaum Foundation, and the Cancer League of Colorado, Inc.
Conflict of interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Hoffman, L.M., Donson, A.M., Nakachi, I. et al. Molecular sub-group-specific immunophenotypic changes are associated with outcome in recurrent posterior fossa ependymoma. Acta Neuropathol 127, 731–745 (2014). https://doi.org/10.1007/s00401-013-1212-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-013-1212-8