
Abstract
Primary lymphoma of the central nervous system (CNS, PCNSL) is a specific diffuse large B cell lymphoma entity arising in and confined to the CNS. Despite extensive research since many decades, the pathogenetic mechanisms underlying the remarkable tropism of this peculiar malignant hematopoietic tumor remain still to be elucidated. In the present review, we summarize the present knowledge on the genotypic and phenotypic characteristics of the tumor cells of PCNSL, give an overview over deregulated molecular pathways in PCNSL and present recent progress in the field of preclinical modeling of PCNSL in mice. With regard to the phenotype, PCNSL cells resemble late germinal center exit IgM+IgD+ B cells with blocked terminal B cell differentiation. They show continued BCL6 activity in line with ongoing activity of the germinal center program. This together with the pathways deregulated by genetic alterations may foster B cell activation and brisk proliferation, which correlated with the simultaneous MYC and BCL2 overexpression characteristic for PCNSL. On the genetic level, PCNSL are characterized by ongoing aberrant somatic hypermutation that, besides the IG locus, targets the PAX5, TTF, MYC, and PIM1 genes. Moreover, PCNSL cells show impaired IG class switch due to sμ region deletions, and PRDM1 mutations. Several important pathways, i.e., the B cell receptor (BCR), the toll-like receptor, and the nuclear factor-κB pathway, are activated frequently due to genetic changes affecting genes like CD79B, SHIP, CBL, BLNK, CARD11, MALT1, BCL2, and MYD88. These changes likely foster tumor cell survival. Nevertheless, many of these features are also present in subsets of systemic DLBLC and might not be the only reasons for the peculiar tropism of PCNSL. Here, preclinical animal models that closely mimic the clinical course and neuropathology of human PCNSL may provide further insight and we discuss recent advances in this field. Such models enable us to understand the pathogenetic interaction between the malignant B cells, resident cell populations of the CNS, and the associated inflammatory infiltrate. Indeed, the immunophenotype of the CNS as well as tumor cell characteristics and intracerebral interactions may create a micromilieu particularly conducive to PCNSL that may foster aggressiveness of tumor cells and accelerate the fatal course of disease. Suitable animal models may also serve as a well-defined preclinical system and may provide a useful tool for developing new specific therapeutic strategies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Historically, lymphoma confined to the central nervous system (CNS) has been a mystery for decades and there are major issues we still face today. This situation is impressively reflected by the nomenclature, which has changed frequently in the 20th century. Initially, Bailey introduced the name “perithelial sarcoma”, but this was later changed many times, including “adventitial sarcoma” and “reticulum cell sarcoma” [22]. These diverse terms acknowledge the peculiar angiotropism of the tumor cells; characteristically, they distend the argyrophilic fibers of the blood vessel walls and may thereby mimic sarcoma, a differential diagnosis more frequently considered by neuropathologists than a hematopoietic tumor, at least in the first half of the 20th century. It was only after the general concept of classifying lymphomas on the basis of a precise morphological description of tumor cells had evolved that neuropathologists noticed the similarity between these intracerebral tumor cells and centroblasts or, less frequently, immunoblasts or centrocytes. This long story came to an end with the development of modern morphological, immunological, molecular cytogenetic, and molecular genetic techniques, which are sensitive enough to cope with the tiny amounts of tissue. The ultimate result is the definition of primary lymphoma of the central nervous system (PCNSL) as a distinct lymphoma entity. Simultaneously, this process provided insights into the pathogenesis of PCNSL and is reflected in the WHO Classification of Tumours series. Specifically, the inclusion of PCNSL in both the WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues and the WHO Classification of Tumours of the Nervous System highlights the unique features of PCNSL and the extraordinary association of malignant hematopoietic cells with the CNS [22, 45].
Definition
Primary CNS lymphoma is defined as malignant lymphoma of the diffuse large B cell (DLBCL) type confined to the CNS [45]. Excluded are lymphomas of the dura (follicular lymphoma, marginal zone lymphoma), intravascular B cell lymphoma, lymphomas with previous or simultaneous systemic disease as well as lymphomas occurring in the setting of immunodeficiency [45]. DLBCL comprise a large and heterogeneous group of mature B cell lymphomas accounting altogether for 40 % of all lymphomas. The issue whether PCNSL are “just” lymphoma, i.e., DLBCL not otherwise specified, in the CNS or better regarded as a distinct lymphoma entity has long been a matter of debate. Similarities between PCNSL and systemic DLBCL and unique features of PCNSL will be compared and contrasted (see below). The mere fact of the existence of a malignant lymphoma confined to an immunoprivileged organ devoid of a classical lymphatic drainage system raises interesting questions regarding the mechanisms of its development, which are of fundamental relevance both scientifically and clinically.
Objectives of a study of PCNSL
Having noticed the hematogenous nature of the tumor cells of PCNSL and their B cell lineage with expression of PAX5, CD19, CD20, CD22, and CD79a, three major issues required investigation to elucidate the pathogenesis of PCNSL: first, accurate identification of the cellular origin of the tumor cells is a conditio sine qua non because lymphoma cells retain characteristic features of their normal counterpart, thus providing a fundamental basis for our understanding of PCNSL. Second, events leading to malignant transformation and sustaining proliferation need to be identified and integrated into a pathogenetically sound concept of PCNSL pathogenesis. Third, the mystery why this lymphoma entity develops in the CNS and, with extremely rare exceptions, remains confined to the CNS needs to be elucidated. The last topic is most challenging and answers are still fragmentary, while enormous progress has been made in our understanding of the histogenetic origin and the identification of molecular events contributing to lymphomagenesis. From these efforts, the concept of PCNSL with unique molecular, biologic, and immune features arose, which is of high clinical and therapeutic relevance, as PCNSL has increased in incidence over the last decades, now accounting for 3–5 % of all CNS tumors [45]. Generally, PCNSL in immunocompetent patients has to be distinguished from PCNSL in immunocompromised patients. While there are morphological and immunophenotypic similarities between PCNSL in both groups of patients, they differ fundamentally in their pathogenesis, which is Epstein Barr virus (EBV)-driven in the latter, but not in the former [50, 61, 62].
Here we focus on the molecular features that determine our current state of knowledge of the biology of PCNSL in immunocompetent patients as a basis for addressing open questions. The ultimate aim is to design specific therapeutic regimens and to improve patients’ outcome. Despite the fact that pathogenetically relevant parameters have been identified, practically none of them have been validated in clinical studies so far. Therefore, at present it is unresolved whether they relate sufficiently strongly to prognostic outcome or treatment.
PCNSL is arrested in terminal B cell differentiation and corresponds to a late germinal center exit B cell
To identify the histogenetic origin of the cells of hematological neoplasms, recapitulation of their differentiation under physiological circumstances can provide a clue, since lymphoma cells retain characteristic features of the non-malignant counterparts from which they likely evolve. Generally, B cell development (Fig. 1) can be regarded as a lifelong maturation process aiming at the recognition of antigens with unique specificity and high affinity. Starting in the bone marrow, where antigen receptor encoding genes are in germline configuration in stem cells, V, D, and J gene segments of the heavy and light chains of the immunoglobulin (IG) genes are assembled, thereby defining a naive B cell, which may now leave the bone marrow. Antigen encounter induces further maturation steps in the germinal center (GC) of secondary lymphoid organs, i.e., lymph nodes and spleen, to increase the binding affinity of the B cell receptor (BCR), the unique receptor for antigen recognition, for its antigen. The molecular correlate of this process is the introduction of somatic mutations into the first 1.5–2 kb of the V region genes of the BCR heavy and light chains. This process of somatic hypermutation (SHM) requires the presence of the specific antigen, antigen presenting cells (APC), T cell help, activity of the enzyme activation induced deaminase (AID), and BCL6. The latter is a master regulator of the GC reaction, which fosters expression of a complex gene program of the GC reaction and represses genes promoting exit of the B cells from the GC [6, 7]. The introduction of somatic mutations into the V region genes of the BCR may either increase or decrease the affinity of the B cell for its antigen and result in selection of the B cell for further rounds of SHM and, finally, exit from the GC or in its apoptosis [56]. Subsequent to SHM, the B cell may undergo IG class switch recombination (CSR). CSR replaces the μ constant region of the BCR with one of the constant regions located downstream to generate diverse antibody classes. This AID-dependent process occurs within the 3- to 5-kb repetitive switch region sequences which are located 5′ of each constant region segment. Differentiation into memory or plasma cells completes B cell differentiation. As all steps of B cell differentiation require DNA strand breaks and, particularly in the GC, include exchange of genetic material at high speed, their failure may yield malignant cells. In fact, erroneous B cell differentiation has been linked to specific lymphoma entities (Fig. 1).

Histogenetic origin of PCNSL. Genotypic and phenotypic characteristics of the tumor cells of PCNSL reveal that they are derived from late GC exit B cells. They correspond to mature B cells arrested in terminal B cell differentiation. aSHM aberrant somatic hypermutation, SHM somatic hypermutation
Regarding B cell differentiation, the tumor cells of PCNSL have reached a mature stage and correspond to GC cells (Fig. 1) [59, 62, 99]. Morphologically, they mostly resemble centroblasts and carry rearranged IG gene segments. The introduction of somatic mutations into the rearranged IG segments [62, 76, 99] proves their participation in a GC reaction. Thus, the tumor cells are antigen-experienced and, in fact, the mutational pattern indicates selection of the tumor cells for an antigen [62, 76, 99]. 60–80 % of PCNSL express the BCL6 protein which is confined to GC B cells [59]. The simultaneous expression of IRF4, which is expressed on a subset of GC and plasma cells, indicates that the tumor cells are on their way to leave the GC. This is supported by their expression of genes more closely related to memory B cells than to GC B cells. Together with cell-surface expression of IGM the combined BCL6 and IRF4 expression indicates that further B cell maturation is impaired. Plasma cell markers (CD38, CD138) are always absent. Important molecular mechanisms underlying these particular features of the geno- and (immuno)phenotype have been elucidated and are outlined below.
Multiple molecular alterations that converge on important pathways regulating B cell activation, immune reactions, and fate foster uncontrolled activation and proliferation
Multiple molecular alterations have been identified by studying PCNSL samples in recent years and have been integrated into a pathogenetic concept explaining the geno- and phenotype of the tumor cells.
Genetic alterations associated with the GC reaction
The fact that PCNSL have experienced a GC reaction with evidence of ongoing SHM [62, 99] provides a clue for our understanding of its pathogenesis (Fig. 1). The GC reaction is a “dangerous” process since it requires DNA strand breaks for subsequent exchange of genetic material. Under physiological conditions, BCL6 tolerizes B cells to DNA damage including breaks. The TP53 gene, a BCL6 target inversely correlated with BCL6, only exceptionally harbors a mutation [17] and is not altered in PCNSL (unpublished observation). During physiological B cell maturation, the GC reaction is tightly regulated in order to minimize genomic instability since derailment can cause oncogenic translocations.
In fact, PCNSL recurrently carry translocations affecting the IG and particularly the BCL6 genes [58, 67, 69]. Indeed, BCL6 translocations were detected in 17, 23, and 47 % of PCNSL [15, 67, 69]. The variation in frequency might be due to the size of the series and the different cut-off levels applied for fluorescence in situ hybridization [58, 67, 69]. Overall, one may expect one-fourth to one-third of PCNSL to harbor a BCL6 break. While intense efforts to identify the translocation partners of the IG genes—with the exception of BCL6—have been unsuccessful so far due to technical limitations, various partner genes (GAPDH, HIST1H4I, HSP90AA1, IGH, IGL, LPP) have been shown to have fused with the promiscuous BCL6 gene [58, 93]. BCL6 gene promoter substitution causes deregulated, constitutive BCL6 activity, which has oncogenic properties [14, 52, 103]. The reciprocally negative feedback of BCL6 and IRF4, a transcription factor expressed in a subset of normal late GC B cells and in plasma cells that physiologically causes extinction in the GC program and, thereby, promotes terminal B cell differentiation, is lost in PCNSL, which strongly express both BCL6 and IRF4 [21]. IRF4 transactivates PRDM1 and both genes drive plasmacytic differentiation [44, 74, 95]. In 19 % of PCNSL, PRDM1 activity is extinguished by deleterious mutations as well as by epigenetic silencing associated with upregulated miR-9 and miR-30b/c [19, 28].
Thus, continued activity of BCL6, a master regulator of the GC reaction modulating B cell activation, differentiation, cell cycle arrest, and apoptosis (for review see [7]), is considered to contribute to keeping the tumor cells of PCNSL in the GC stage. In PCNSL, the process of SHM is not confined to IG and BCL6 genes, but also involves other genes implicated in tumorigenesis. This active process of aberrant SHM may either activate or inactivate oncogenes and tumor suppressor genes including PAX5 (60 %), TTF (70 %), CMYC (60 %), and PIM1 (50 %) [68]. The fixed IGM/IGD phenotype of the tumor cells is in part due to miscarried CSR attempts of the tumor cells during which the sμ region was deleted [66].
In addition to translocations, gains and losses of genetic material are recurrent in PCNSL as evidenced by genome-wide single-nucleotide polymorphism chip analysis and FISH analyses. Overall, gains are more frequent than losses affecting 18q21.33-q23 (43 %), chromosome 12 (26 %), and 10q23.21 (21 %), while losses were detected in 6q21 (52 %), 6p21 (37 %), 8q12.1–q12.2 (32 %), and 10q23.21 (21 %) [94].
Epigenetic alterations
Epigenetic alterations may also contribute to PCNS pathogenesis. This includes epigenetic silencing by DNA methylation. Several genes including DAPK (84 %), CDKN2A (75 %), MGMT (52 %), and RFC (30 %) are targeted by DNA hypermethylation and potentially may have therapeutic relevance [16, 27, 34]. In a subgroup of elderly PCNSL patients with methylated MGMT promoter, temozolomide monotherapy appeared to be therapeutically effective [49]. However, studies have not yet been performed in large numbers of patients. Genome-wide array-based DNA methylation profiling identified 194 genes as differentially methylated between PCNSL and normal hematopoietic controls with a significant enrichment of polycomb target genes and genes with promoters showing a high CpG content [80]. Comparison of methylation profiles of PCNSL and systemic DLBCL, however, did not reveal major differences [80].
Our current knowledge of miRNA expression in PCNSL is still limited. Two studies on PCNSL samples reported a differential pattern of miRNA expression in PCNSL, however, with virtually no overlap. In a study of 11 PCNSL, the miRNAs miR-9 and miR-30b/c, which are associated with terminal B cell differentiation, were upregulated in PCNSL [28]. In another study of 9 PCNSL, miR-17-5p, which targets the proapoptotic E2F1 gene, and miR-10a, were expressed at significantly higher levels in PCNSL than in nodal DLBCL [84]. The analysis of cerebrospinal fluid (CSF) from 23 patients with PCNSL showed miR-21, miR-19, and miR-92 to be significantly elevated compared to controls with inflammatory CNS disorders [2]. Thus, approaches differing in materials and methodology may account, at least in part, for these discrepancies.
Activation of pathways fostering tumor cell survival
Constitutive activation of the nuclear factor (NF)-κB pathway in PCNSL (Fig. 2) was evidenced by comparative studies of genes upstream and downstream of the NF-κB complex as well as of NF-κB complex genes in PCNSL and non-malignant tonsillar GC B cells. Interestingly, genes of this pathway (BAX, BCLXL, BCL2, MALT1, CARD9, CARD10, CARD11, CARD14, CCND2, CFLIP, RELA, RELB, NFKB1, NFKB2, IRF4) were more prominently expressed in PCNSL [20]. These alterations could be explained by mutations targeting components of several regulatory pathways upstream of the NF-κB complex including the toll-like receptor (TLR), the BCR signaling pathway as well as its BCR pathway target, the BCM complex, are affected by mutations (Fig. 2). The BCR complex consists of the IG heavy and light chains as well as of CD79A and CD79B subunits. Its expression is indispensable for B cell survival. BCR signaling induces differentiation, proliferation, and apoptosis of B cells. Crucial components of genes of the BCR signaling cascade are altered by somatic mutations in PCNSL (44 %) causing deregulated BCR signaling, however, not abolishing BCR signaling [64]. Mutations affected the SHIP (25 %), CD79B (20 %), CBL (4 %), and BLNK (4 %) genes and thus altered genes with tumor suppressor (SHIP, CBL, BLNK) or protooncogene (CBL) function [64]. In contrast, the SPIB gene, an Ets family transcription factor which interacts with IRF4 to amplify the NF-κB signaling required for full BCR signaling and is also important for the GC reaction [43], was not targeted by somatic mutations in PCNSL (unpublished observation).

Genetic alterations involved into the pathogenesis of PCNSL converge on important pathways for B cell signaling. A plethora of genes are affected by point mutations, amplifications, translocations, and deletions. Other genes are deregulated by yet unknown mechanisms. They target the signaling cascades of the BCR pathway, NF-κB pathway and their targets, i.e., transcription factors. Worthy of note is that many genes described as being highly mutated in other tumors, e.g., TP53 or PTEN, or switched off by mutations in systemic DLBCL, like TNFAIP3, do not play a major role in PCNSL. These observations indicate a different selection pressure in the CNS, their specialized and unique localization. BCR, B cell receptor; IgA, CD79A; IgB, CD79B; TLR toll-like receptor; CR chemokine receptor
Signals from the BCR are transmitted to the BCM complex consisting of three components, i.e., BCL10, MALT1, and CARD11, which is also subject to various alterations in PCNSL (Fig. 2). Amplification of MALT1 may underly the prominent expression of its gene product in PCNSL, which also express BCL10 [20, 67, 69, 81, 102]. Furthermore, 16 % of PCNSL harbored oncogenically activating mutations of the CARD11 gene, which encodes a scaffold protein required for antigen receptor-induced NF-κB activation [65]. Thus, in addition to enhanced signaling from the BCR, its target is altered, thereby further boosting NF-κB activation.
Moreover, the TLR pathway is also deregulated in 50 % of PCNSL due to mutations of the MYD88 gene, the central adaptor protein (Fig. 2) [60]. These data were recently confirmed in an independent series [33]. In 36 % of affected PCNSL, mutations corresponded to a leucine to proline exchange at position 265 (L265P) [60], which is an oncogenically activating mutation [72]. 40 % of the PCNSL with this L265P mutation concomitantly harbored a CARD11 mutation [60], allowing synergism to further enhance NF-κB activity. On the other hand, CARD11 mutations were not correlated with mutations in genes of the BCR pathway in PCNSL [64] indicating potentially complementary effects of these mutations on NF-κB activity.
In addition to the BCR and TLR, which activate the classical pathway, BAFF receptor mediated stimulation may sustain NF-κB activity via activation of the non-canonical pathway. In PCNSL, BAFF receptor expressing tumor cells may react to BAFF produced by astrocytes [48].
Collectively, several potent pathways upstream of the NF-κB complex are altered which may contribute either alone or in synergy to the activation of the NF-κB pathway. The high constitutive NF-κB activity in PCNSL also explains the prominent IRF4 expression by the tumor cells even in the absence of IRF4 translocations (unpublished observation), because IRF4 is an NF-κB target gene. Ultimately, these various alterations converge to induce and sustain a proliferative, non-apoptotic phenotype of the B cells, which exhibit a remarkably increased proliferation rate reaching a MIB-1 index of up to 70–90 % [21], and may in part explain the poor prognosis of patients with PCNSL.
Role of the CNS microenvironment in PCNSL
Deciphering the interactions between the three major players in the CNS with PCNSL, i.e., the malignant B cells, resident cells of the CNS, and immune cells recruited to the brain, will provide a clue to our understanding of the pathogenesis of PCNSL. In nodal lymphomas, gene expression profiling studies have identified a lymph node signature which reflected the character of the non-malignant cells in the tumor with genes of the extracellular matrix deposition and histiocytic infiltration and which were associated with a favorable prognosis [51]. In PCNSL, the role of the target organ is even more challenging, as the immune reactions to the malignant B cells in an immunoprivileged organ represent a special scenario. Its elucidation will lead us beyond the disease of PCNSL and be of fundamental neuroimmunological interest. Thus, at present, understanding the mechanisms underlying the selective tropism and confinement of this lymphoma entity to the CNS is the most challenging task in decoding the mystery of lymphoma confinement to the CNS.
B cell homing to the CNS and intracerebral migration
So far, the question whether B cells home to the CNS in a benign or malignant state has not been answered. B cells which have been recruited to the CNS in the course of an immune reaction, most likely in response to a pathogen, may have persisted for extended periods and eventually may have transformed while residing inside the brain. On the other hand, B cells may also have transformed outside the CNS, e.g., during a GC reaction in a secondary lymphoid organ, and entered the CNS thereafter. Generally, both of these mechanisms are conceivable. Regarding the latter hypothesis, homing of a malignant B cell exclusively to the CNS cannot be explained. So far, neither a cell adhesion molecule nor a chemokine pattern predicting B cell homing selectively to the CNS was identified. PCNSL express cell adhesion molecules similar to nodal DLBCL including α3β1 and α4β1-1 and ICAM-1 [5, 75] (Fig. 3). Interestingly, tumor cells loosely infiltrating the brain tissue showed lower αLβ2 integrin than compact cell clusters suggesting that this integrin is related to the infiltration pattern of PCNSL within the brain parenchyma [75]. Binding of osteopontin expressing tumor cells to CD44, which is expressed by astrocytes [1, 36, 96], microglia [55, 96], cerebral endothelial cells [86], and infiltrating T cells, may promote migration and brain tissue invasion of the lymphoma cells [104] (Fig. 3).

Interaction between the major players in the CNS in PCNSL, i.e., tumor B cells, resident brain cells as well as reactive inflammatory bystander cells. Via their expression of chemokines (CCL5), chemokine receptors (CXCR4, CXCR5), cytokine receptors (B cell activating factor, BAFF-R), cell adhesion molecules (lymphocyte function-associated antigen 1, LFA-1), the glycoprotein osteopontin, and—in some, but not all cases—MHC class II antigen, tumor B cells may interact with their corresponding ligand on astrocytes (CCL5/CCR5, BAFF-R/BAFF), on microglial cells (CCL5/CCR5, osteopontin/CD44, MHC class II antigen), on reactive T cells (osteopontin/CD44), and on endothelial cells [LFA-1/intercellular adhesion molecule-1 (ICAM-1), CXCR4/CXCL12, CXCR5/CXCL13, osteopontin-CD44, MHC class II antigen]. These interactions may contribute to the morphological hallmarks of intracerebral lymphoma spread, i.e., formation of perivascular tumor cell cuffs via LFA-1/ICAM-1, CXCR4/CXCL12, and CXCR5/CXCL13 and diffuse brain infiltration via CCL5/CCR5, BAFF-R/BAFF, and osteopontin/CD44 interactions, respectively
While under physiologic conditions, the CNS is characterized by an immunologically downregulated phenotype, a complex neuroimmune network is readily activated in the CNS under pathological circumstances, be it of inflammatory or tumorous nature. This is also the case for PCNSL, in which activated CD4 and CD8 T cells, reactive B lymphocytes, and macrophages are recruited to the brain, where they are associated with the lymphoma cells. In addition, cerebral endothelial cells, astrocytes, and microglia are strongly activated and respond in a cell-type specific manner to the malignant cells, thus, allowing cell-type specific reactions with the tumor cells (Fig. 3). While the tumor cells express CXCR4 and CXCL12, CXCR5 and CXCL13, astrocytes and microglia upregulated CXCL12, CCR5, and CCR6, endothelium expressed CXCL12 and CXCL13, and T cells expressed CCR5 and CCR6 [12]. In addition to diffusely infiltrating the brain, there is a tendency for CD8 T cells to accumulate in perivascular areas, which was correlated with CXCL9 expressed by perivascular macrophages and pericytes [78]. Binding of CXCL9 to CXCL12 can enhance migration of CXCR4+CXCR3+ CD8 T cells as well as CXCR4+ tumor B cells [12, 78]. This expression pattern of chemokines may contribute to the angiotropism of the malignant B cells. It has also been claimed that angiotropism of the tumor cells is fostered by IL-4 expressed by cerebral endothelial cells [88]. However, it is unlikely that a single mediator may underly this characteristic architecture of PCNSL. In this regard it is important to recognize that anatomy of the neurovascular unit is much more complex.
The neurovascular unit consists of three compartments, which are separated by distinct basement membranes, i.e., the vascular wall, the perivascular Virchow-Robin space, and the juxtavascular parenchyma (neuropil) (for review see [47]). To date, most of our knowledge on recruitment, homing, pattern of distribution of lymphocytes in the CNS and their restimulation in the CNS is based on studies of T cells in autoimmune and infectious CNS inflammation, while functional studies on malignant B cells have not been reported. In the perivascular space, where macrophages at various activation states reside, lymphocytes readily establish contact with APC which present antigen and stimulate incoming lymphocytes [77]. After passage of the basement membrane of cerebral blood vessel endothelial cells, lymphocytes residing in the perivascular space may interact with perivascular macrophages [8]. In the case of T cells in CNS inflammation, these cells are in search of their specific antigen [4]. Antigen recognition is required for subsequent invasion of the CNS parenchyma. In the absence of antigen, T cells remain confined to the perivascular space [77]. Hypothesizing an analogous process for malignant B cells, their confinement and preferential accumulation in the perivascular space may be due to lack of antigen-specific interactions. Such a scenario may explain the accumulation of malignant B cells within perivascular cuffs after their injection into the brain of rodents in non-syngeneic models [42]. On the other hand, it is also conceivable that the presence of perivascular APC, which also reside in the choroid plexus, beneath the ependyma [47], may create a micromilieu particularly convenient for the tumor cells of PCNSL, fostering their activation and contributing to their sustained proliferation. In addition, the tumor cells may also contribute to a favorable micromilieu, thereby affecting proliferation, the pattern and extent of intracerebral spread, and may also influence the anti-tumor immune response. Regarding this issue, only the tip of the iceberg has yet been addressed, as in addition to astrocytes, microglia/macrophages, and endothelial cells it is likely that other cell populations of the CNS including neurons, oligodendrocytes, ependymal cells, and choroid plexus epithelial cells are also likely to contribute to the immune response and the interactions with the tumor cells. So far, emphasis has concentrated on the expression of immunologically relevant cell-surface molecules.
Soluble mediators in PCNSL
In addition, it is likely that a plethora of soluble mediators interacting in a complex, finely tuned network play an important role in the interactions of the malignant B lymphocytes and their neighbors. Molecular alterations that are important for malignant transformation may also play a role in this regard. For instance, MYD88 mutations [60] may alter the cytokine milieu by inducing IL-6 and IL-10 secretion of tumor cells via activation of IRAK4. Furthermore, BCL6 modulates a number of signaling pathways involved in the response to cytokines and chemokines including IL-6, and interleukin receptors: IL-10RA and IL-10RB, and STAT family members, e.g., are targets of BCL6 [6]. Interestingly, PCNSL patients were reported to differ from healthy controls in their IL-101082 genotype distribution [79]. In addition, elevated IL-10 levels were recently reported in the CSF of PCNSL patients as compared to other tumors and inflammatory CNS disorders [89]. However, these data are at variance with a number of studies that detected IL-10 in the CSF in pathological CNS conditions, in particular in bacterial meningitis, but also in other neuroinflammatory disorders [31, 32, 38, 46, 53, 100, 101]. Since IL-10 is well-known to counterbalance pro-inflammatory immune reactions in the course of CNS inflammation and has been demonstrated to play an important role in this scenario [23, 24, 29, 30, 85, 92, 98], the recent report by Rubenstein et al. [89] is still in contradiction. Taken the data from various studies together, it is highly unlikely that CSF IL-10 levels can be regarded as PCNSL specific, in particular considering the complex neuroimmunological scenario in PCNSL that makes a single mediator as diagnostic parameter very unlikely.
Impact of MHC class I and II molecule expression on the tumor cells of PCNSL
Remarkably, 73 % of PCNSL were affected by heterozygous and even homozygous loss or partial uniparental disomies of the chromosomal region 6p21.32 harboring the MHC class II encoding genes HLA-DRB, HLA-DQA, and HLA-DQB [94]. Correspondingly, 55 and 46 % of PCNSL had lost expression of HLA-A and HLA class II, respectively [11]. Similar to PCNSL, HLA class I and II molecules were not expressed on the tumor cells of testicular lymphoma, another DLBCL manifesting in an immunoprivileged organ [11]. In testis lymphoma a low level of HLA-DR mRNA was associated with significantly lower numbers of infiltrating CD3 T cells [82]. Of clinical importance is the fact that MHC class II-negative systemic DLBCL were correlated with a poor outcome [83]. Interestingly, this may also hold true for PCNSL: Patients with reactive perivascular infiltrates (RPVI) exhibited a significantly better overall survival than patients with RPVI-negative PCNSL, particularly among patients treated with high-dose methotrexate chemotherapy [78]. These data suggest that loss of MHC molecules may provide a survival advantage and may allow the tumor cells to escape from the (neuro)immune response.
Preclinical models for PCNSL
The observed clinically relevant data necessitate functional studies addressing the interaction between the tumor cells and the various cell populations of the nervous system and the immune system. So far, in vitro studies have been precluded since cell lines could not be established from PCNSL and appropriate animal models were not available. In the past, immunodeficient rodents were used which received human PCNSL cells either intracerebrally or outside the brain; however, these models do not allow adequate immunological studies. Several groups have implanted human MC116 B cells into the basal ganglia of athymic nude rats, 84 % of which developed a tumor [39, 71, 97]. This rate could be increased to 92 % by pretreatment with cyclophosphamide [97]. Furthermore, correlation between MRI and histology was poor [97]. While i.v. rituximab proved highly effective, methotrexate was only minimally effective therapeutically as evidenced by evaluation of posttreatment MRI scans [39]. However, the MC116 cell line is, like Raji cells, which grow readily in the brain of athymic nude mice [42], a Burkitt lymphoma cell line [73]; since Burkitt lymphoma cells are entirely different from those of PCNSL [59] and DLBCL, they are irrelevant for investigation of PCNSL. The same holds true for EBV+ human lymphoblastoid B cell lines injected into athymic nude rats, which were used for radiation therapy studies that demonstrated that irradiation sensitized cells to nucleoside analogs-induced apoptosis [87]. Mineo et al. [57] injected the 38C13 large B cell line of C3H murine origin into the basal ganglia of C3H mice. Local intracerebral application of rituximab at day 1 after injection of the tumor cells prevented cerebral tumor growth in 67 % (8/12) of mice [57]. Basically, these experiments are interesting. However, this regimen does not target established PCNSL and, thus, is clinically irrelevant. Taken together, these various models do not parallel human PCNSL appropriately.
Murine PCNSL as paradigm for human PCNSL
In addition to the requirement of a syngeneic system, lymphoma cells optimally should exhibit major characteristics of PCNSL, preferably exhibiting GC B cell features, since the stage of differentiation is an important parameter determining the ensuing immune reaction. A major step forward was the establishment of a murine model in which the A20.IIA cell line was introduced into the brain of syngeneic mice resulting in lethal lymphoma [26]. Intracerebral growth of the malignant B cells was associated with recruitment of CD4 and CD8 T cells, reactive B cells and with an increase in CD11b+ mononuclear cells and CD11c+ cells in the tumor. Leukocytes freshly isolated from the brain of tumor-bearing mice did not produce cytokines, but could be induced to do so by ex vivo stimulation with anti-CD3/CD28 monoclonal antibodies in the presence of A20.IIA cells yielding concomitant Th1/Th2/TH17 profiles (IL-2, IL-4, IL-17, IFN-γ, GM-CSF) [26]. Athymic nude mice succumbed significantly earlier to cerebral or splenic lymphoma than their immunocompetent BALB/c counterpart indicating that T cells play a role in the response to the lymphoma [26]. This model proved valuable in addressing therapeutic regimens directed specifically at individual features of the tumor cells. Interestingly, TLR9 expressing A20.IIA cells could be targeted by CpG-ODNs, a TLR9 agonist, and showed tumor regression when injected into the tumor 7 days after implantation [9]. Since this procedure is rather early after transplantation before onset of clinical symptoms, its rationale in the treatment of PCNSL patients with established tumor burden is debatable. Transfection of A20.IIA cells with human CD20 rendered murine lymphoma cells susceptible to ublituximab, which was injected into the cerebral lymphoma 4–7 days after implantation [10].
However, A20.IIA cells express IGG [40] and, thus, are more mature than IGM+ PCNSL, in which lack of CSR is a hallmark due to specific molecular alterations [66].
We have recently developed a syngeneic murine model of PCNSL using the BAL17 cell line, a mature tumor B cell derived from a BALB/c mouse [40, 54, 63]. To the best of our knowledge, at present there is no mouse cell line available, which resembles PCNSL more closely. BAL17 cells express CD19 and B220 and carry functionally rearranged IG genes that express IGM without having performed CSR [63]. Using the Bethesda classification of lymphoid neoplasms in mice [70], BAL17-induced lymphoma is characterized as murine DLBCL of the centroblastic-immunoblastic type. This model recapitulates major features of PCNSL and allows investigation of the in vivo interactions between the malignant B cells and cells in their target organ. Interestingly, after transplantation of BAL17 cells into the frontal lobe of immunocompetent BALB/c mice, 100 % of the animals developed PCNSL that ran a lethal course with a particular tropism of the lymphoma cells for the inner and outer ventricular system and the basal ganglia (Fig. 4). The angiocentric growth pattern, a hallmark of human PCNSL, was also reproduced by BAL17 cells (Fig. 4) indicating that the perivascular Virchow-Robin space may provide a particularly fertile microenvironment for the lymphoma cells [63]. This local position, however, may be a unique feature for B cells, be they both malignant or not. In various inflammatory (autoimmune and infectious) CNS disorders in humans and mice, B cells recruited to the CNS mainly stay in the perivascular space and do not spread throughout the brain as typically observed for T cells [91]. The preferential residence in the perivascular space may also indicate a strong interaction of the malignant B cells with components of the blood–brain barrier, particularly cerebral endothelial cells, which have upregulated MHC class I and II antigens, ICAM-1, and VCAM-1 [63]. Interestingly, activation of resident brain cells was locally confined to areas of lymphoma infiltration, where microglia and astrocytes upregulated ICAM-1, MHC class I and II antigens, and GFAP, respectively [63]. CD4 and CD8 T cells were intermingled with the tumor cells [63]. So far, the precise function of T cells in murine PCNSL is still unknown. In the A20.II CNS lymphoma model, T cells recruited to the brain were mainly of the regulatory type [26], which raises the hypothesis of a permissive T cell response. Interestingly, repeated isolation and re-transplantation of BAL17 cells into the brains of BALB/c mice accelerated intracerebral tumor growth and significantly shortened the time to death [63]. This poorer clinical course was attributed to an altered genotype and phenotype of the lymphoma cells. Alterations predominantly affected genes involved in the regulation of apoptosis, including Sp110, Birc3, Xaf1, and Ncf1 as well as genes of the JAK/STAT pathway (Il-9r, Stat1, Jak2) and immune response genes (Tlr-1, Ccl5, Il-1b, Cd74, H-2), all of which were upregulated [63]. The increased expression of immune response genes such as Ccl5 may facilitate intracerebral spread of the lymphoma cells by fostering their interactions with microglia and astrocytes, which, in human PCNSL express the corresponding ligand CCR5 [12]. The increased TLR1 expression may foster tumor cell proliferation by stimulation of the NF-κB pathway. Correspondingly, the re-transplanted lymphoma cells expressed higher levels of MHC class I and II antigens [63], indicating their enhanced activation, which may also facilitate interactions with cells of the nervous and the immune system. These data support the hypothesis that the microenvironment of the CNS provides a fertile environment for B lymphoma cells and may even induce a more malignant phenotype of the tumor cells. Thus, the syngeneic BAL17-PCNSL model recapitulates major features of human PCNSL and provides a useful tool for analyzing interactions of the tumor cells with individual components of the very special microenvironment of the CNS.

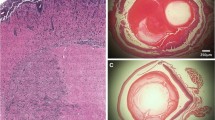
Neuropathology of BAL17-induced primary CNS lymphoma in BALB/c mice at day 28 after intracerebral transplantation. a The fronto-parietal leptomeninges are heavily infiltrated by densely packed lymphoma cells extending the subarachnoid space (asterisks). From here, tumor cells have invaded the superficial cortical layer. The dotted line denotes the cortical surface. Note the angiocentric growth pattern of lymphoma cells in the brain parenchyma (arrows) in addition to diffuse, sheet-like lymphoma growth. H&E staining; original magnification ×400. b Tumor cells express the pan-B-cell marker CD19. The arrow indicates the angiocentric growth pattern. Small clusters of lymphoma cells are diffusely scattered throughout the brain parenchyma. Immunohistochemistry with rat anti-mouse CD19 and slight counterstaining with hemalum; original magnification ×400. c The proliferative activity of the lymphoma cells is very high (>90 %). At day 28, dense sheets of lymphoma cells have infiltrated the leptomeninges. Immunohistochemistry with rabbit anti-mouse Ki-67 and slight counterstaining with hemalum; original magnification ×200
Future directions
In recent years, remarkable progress has been made in our understanding of the nature of PCNSL. Consensus has been achieved that PCNSL are a specific entity exhibiting both unique features and overlap with molecular characteristics of other DLBCL outside the CNS. For instance, in systemic DLBCL, expression of the transcription factor FOXP1 is associated with a particularly poor clinical outcome [3]. PCNSL are also characterized by FOXP1 overexpression [18]. However, in contrast to systemic DLBCL, PCNSL exhibit a more complex alteration with a preferential overexpression of the FOXP1 isoforms 3 and 9 together with a downregulation of the normal isoform 1 [18]. The functional consequences of this kind of FOXP1 alteration is still unknown, in particular, whether it contributes to the impaired CSR in PCNSL analogous to mice which transgenically express this transcription factor and which are impaired in their CSR switch [90].
Progress has been achieved with respect to all three major pending issues specified here. Regarding the question as to the histogenetic origin, the tumor cells were identified as exhibiting a late GC exit phenotype [59, 62, 67, 68]. At present, however, our understanding of the differentiation state of GC B cells with their genotypic and phenotypic characteristics is in the process of diversification leading to a differentiation into functionally different subtypes which may give rise to distinct malignancies. The further narrowing down of the cell of origin of PCNSL will be the next step that is likely to be of relevance for our understanding of its pathogenesis.
Despite the fact that many molecular alterations at the level of individual genes, epigenetics, converging on functionally relevant pathways have been identified in PCNSL, the deciphering of further molecular alterations in PCNSL will continue to be in the focus of research to gain a deeper insight into its molecular pathogenesis. This will include a thorough analysis of the genome by whole exome sequencing, studies of miRNA profiles, proteomics, and metabolomics. In these fields, studies are either not available or have been confined to only limited number of cases. For example, our current knowledge of miRNA expression in PCNSL is limited to the analysis of 20 PNCSL in two separate studies using different approaches [28, 84] and, thus, the respective results are not comparable. So far, whole exome sequencing has been reported for four cases of PCNSL in which sufficient tumor DNA and matched blood DNA were available [33]. Overall, 1,678 somatic mutations involving 1,424 genes were detected [33]. After Sanger sequencing, five somatic mutations in three genes, i.e., MYD88, PIM1, and TBL1XR1 were reconfirmed [33]. These observations confirm our previous data on MYD88 and PIM1 mutations in PCNSL [60, 68] and reveal one novel mutation affecting the TBL1XR1 gene [33]. TBL1XR1 plays a regulatory role in the NF-κB pathway and Wnt-mediated transcription and it has been suggested that it is inactivated as a tumor suppressor gene in PCNSL [33]. In order to allow representative conclusions, many more cases need to be analyzed.
The prognostic role of most of the molecular alterations identified is still unknown. So far, a prognostic relevance has been identified only for a very limited number of parameters including a loss of copy numbers on chromosome 6q in 22 PCNSL patients [81]. This was recently confirmed in another study of 29 homogenously treated patients, in which CDKN2A homozygous deletion also was associated with shorter progression-free survival and with overall survival [33]. Methylation of the RFC promoter was associated with a lower complete remission rate to high dose methotrexate-based chemotherapy; however, this observation was based on data obtained in 18 patients [27].
The unfavorable outcome of PCNSL might also be due to the frequent triple expression of the MYC, BCL2, and BCL6 proteins in PCNSL, and, moreover, the absence of a MYClowBCL2low subgroup [13]. The latter has been shown to be associated with a favorable prognosis in systemic DLBCL [35, 37, 41]. In a series of 50 PCNSL, we identified 82 % of PCNSL as MYChighBCL2high by immunohistochemistry [13], a fraction significantly exceeding that in systemic DLBCL [35, 37, 41]. Interestingly, none of the 50 PCNSL lacked both MYC and BCL2 expression [13]. In contrast to protein expression, only 8 % (4/49) of PCNSL showed a MYC break, thus revealing a striking discrepancy between the high level of MYC protein expression and the scarcity of MYC translocations [13]. In this regard, one may speculate that BCR mediated signaling may foster MYC expression [25] also in PCNSL. Interestingly, in PCNSL, MYC overexpression was significantly correlated with the extremely high proliferative activity [13].
All of these potentially clinically relevant observations still await confirmation in a large group of patients. The general paucity of evidence of clinical relevance of molecular parameters can be explained by the lack of large-scale prospective therapeutic studies encompassing a series of patients sufficient in number to allow the identification of prognostically relevant parameters. So far, studies have been either performed retrospectively, frequently in homogenously treated patients or have been restricted to a limited number of patients much below the size of groups of extracerebral DLBCL, and have yielded controversial data or lacked statistical power. Since PCNSL is a rare disease, this problem should be overcome by intense cooperation between the various groups in the field. They should join their efforts and perform international multicenter clinical studies embedded in a well-defined research program.
The most recent progress providing an eminent step forward is, from our point of view, the establishment of a reliable and well-reproducible syngeneic animal model, which allows us to precisely analyze the impact of the microenvironment and the cross-talk of the tumor cells of PCNSL with the other players in the CNS, i.e., cells of the immune system that constitute the inflammatory reactive infiltrate and resident cells of the CNS that are activated to interact with the lymphoma cells via cell-surface molecules and/or soluble mediators. Deciphering the role of the multiple factors involved in this complex scenario will provide a major future challenge ultimately destined to contribute to a thorough and targeted therapy.
References
Alldinger S, Fonfara S, Kremmer E, Baumgärtner W (2000) Up-regulation of the hyaluronate receptor CD44 in canine distemper demyelinated plaques. Acta Neuropathol 99:138–146
Baraniskin A, Kuhnhenn J, Schlegel U et al (2011) Identification of microRNAs in the cerebrospinal fluid as marker for primary diffuse large B-cell lymphoma of the central nervous system. Blood 117:3140–3146
Barrans SL, Fenton JA, Banham A, Owen RG, Jack AS (2004) Strong expression of FOXP1 identifies a distinct subset of diffuse large B-cell lymphoma (DLBCL) patients with poor outcome. Blood 104:2933–2935
Bartholomäus I, Kawakami N, Odoardi F et al (2009) Effector T cell interactions with meningeal vascular structures in nascent autoimmune CNS lesions. Nature 462:94–98
Bashir R, Coakham H, Hochberg F (1992) Expression of LFA-1/ICAM-1 in CNS lymphomas: possible mechanism for lymphoma homing into the brain. J Neurooncol 12:103–110
Basso K, Dalla-Favera R (2010) BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv Immunol 105:193–210
Basso K, Dalla-Favera R (2012) Roles of BCL6 in normal and transformed germinal center B cells. Immunol Rev 247:172–183
Bechmann I, Priller J, Kovac A et al (2001) Immune surveillance of mouse brain perivascular spaces by blood-borne macrophages. Eur J Neurosci 14:1651–1658
Ben Abdelwahed R, Cosette J, Donnou S et al (2013) Lymphoma B-cell responsiveness to CpG-DNA depends on the tumor microenvironment. J Exp Clin Cancer Res 32:18
Ben Abdelwahed R, Donnou S, Ouakrim H et al (2013) Preclinical study of ublituximab, a glycoengineered anti-human CD20 antibody, in murine models of primary cerebral and intraocular B-cell lymphomas. Invest Ophthalmol Vis Sci 54:3657–3665
Booman M, Douwes J, Glas AM et al (2006) Mechanisms and effects of loss of human leukocyte antigen class II expression in immune-privileged site-associated B-cell lymphoma. Clin Cancer Res 12:2698–2705
Brunn A, Montesinos-Rongen M, Strack A et al (2007) Expression pattern and cellular sources of chemokines in primary central nervous system lymphoma. Acta Neuropathol 114:271–276
Brunn A, Nagel I, Montesinos-Rongen M et al (2013) Frequent triple-hit expression of MYC, BCL2, and BCL6 in primary lymphoma of the central nervous system and absence of a favorable MYClowBCL2low subgroup may underlie the inferior prognosis as compared to systemic diffuse large B cell lymphomas. Acta Neuropathol doi. doi:10.1007/s00401-00013-01169-00407
Butler MP, Iida S, Capello D et al (2002) Alternative translocation breakpoint cluster region 5′ to BCL-6 in B-cell non-Hodgkin’s lymphoma. Cancer Res 62:4089–4094
Cady FM, O’Neill BP, Law ME et al (2008) Del(6)(q22) and BCL6 rearrangements in primary CNS lymphoma are indicators of an aggressive clinical course. J Clin Oncol 26:4814–4819
Chu LC, Eberhart CG, Grossman SA, Herman JG (2006) Epigenetic silencing of multiple genes in primary CNS lymphoma. Int J Cancer 119:2487–2491
Cobbers JM, Wolter M, Reifenberger J et al (1998) Frequent inactivation of CDKN2A and rare mutation of TP53 in PCNSL. Brain Pathol 8:263–276
Courts C, Brunn A, Montesinos-Rongen M et al (2009) Preferential expression of truncated isoforms of FOXP1 in primary central nervous system lymphoma. J Neuropathol Exp Neurol 68:972–976
Courts C, Montesinos-Rongen M, Brunn A et al (2008) Recurrent inactivation of the PRDM1 gene in primary central nervous system lymphoma. J Neuropathol Exp Neurol 67:720–727
Courts C, Montesinos-Rongen M, Martin-Subero JI et al (2007) Transcriptional profiling of the nuclear factor-kappaB pathway identifies a subgroup of primary lymphoma of the central nervous system with low BCL10 expression. J Neuropathol Exp Neurol 66:230–237
Deckert M, Brunn A, Montesinos-Rongen M, Terreni MR, Ponzoni M (2013) Primary lymphomas of the central nervous system—a diagnostic challenge. Hematol Oncol. doi:10.1002/jon.2087
Deckert M, Paulus W (2007) Malignant lymphomas. In: Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) WHO classification of tumors pathology ans genetics of tumours of the nervous System, 4th edn. IRAC, Lyon, pp 188–192
Deckert M, Soltek S, Geginat G et al (2001) Endogenous interleukin-10 is required for prevention of a hyperinflammatory intracerebral immune response in Listeria monocytogenes meningoencephalitis. Infect Immun 69:4561–4571
Deckert-Schlüter M, Buck C, Weiner D et al (1997) Interleukin-10 downregulates the intracerebral immune response in chronic toxoplasma encephalitis. J Neuroimmunol 76:167–176
Dominguez-Sola D, Victora GD, Ying CY et al (2012) The proto-oncogene MYC is required for selection in the germinal center and cyclic reentry. Nat Immunol 13:1083–1091
Donnou S, Galand C, Daussy C et al (2011) Immune adaptive microenvironment profiles in intracerebral and intrasplenic lymphomas share common characteristics. Clin Exp Immunol 165:329–337
Ferreri AJ, Dell’Oro S, Capello D et al (2004) Aberrant methylation in the promoter region of the reduced folate carrier gene is a potential mechanism of resistance to methotrexate in primary central nervous system lymphomas. Br J Haematol 126:657–664
Fischer L, Hummel M, Korfel A, Lenze D, Joehrens K, Thiel E (2011) Differential micro-RNA expression in primary CNS and nodal diffuse large B-cell lymphomas. Neuro Oncol 13:1090–1098
Frei K, Lins H, Fontana A (1994) Production and function of IL-10 in the central nervous system. Schweiz Arch Neurol Psychiatr 145:30–31
Frei K, Lins H, Schwerdel C, Fontana A (1994) Antigen presentation in the central nervous system. The inhibitory effect of IL-10 on MHC class II expression and production of cytokines depends on the inducing signals and the type of cell analyzed. J Immunol 152:2720–2728
Frei K, Nadal D, Pfister HW, Fontana A (1993) Listeria meningitis: identification of a cerebrospinal fluid inhibitor of macrophage listericidal function as interleukin 10. J Exp Med 178:1255–1261
Gallo P, Sivieri S, Rinaldi L et al (1994) Intrathecal synthesis of interleukin-10 (IL-10) in viral and inflammatory diseases of the central nervous system. J Neurol Sci 126:49–53
Gonzalez-Aguilar A, Idbaih A, Boisselier B et al (2012) Recurrent mutations of MYD88 and TBL1XR1 in primary central nervous system lymphomas. Clin Cancer Res 18:5203–5211
Gonzalez-Gomez P, Bello MJ, Arjona D et al (2003) CpG island methylation of tumor-related genes in three primary central nervous system lymphomas in immunocompetent patients. Cancer Genet Cytogenet 142:21–24
Green TM, Young KH, Visco C et al (2012) Immunohistochemical double-hit score is a strong predictor of outcome in patients with diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 30:3460–3467
Haegel H, Tölg C, Hofmann M, Ceredig R (1993) Activated mouse astrocytes and T cells express similar CD44 variants. Role of CD44 in astrocyte/T cell binding. J Cell Biol 122:1067–1077
Horn H, Ziepert M, Becher C et al (2013) MYC status in concert with BCL2 and BCL6 expression predicts outcome in diffuse large B-cell lymphoma. Blood 121:2253–2263
Ishiguro A, Suzuki Y, Inaba Y, Komiyama A, Koeffler HP, Shimbo T (1996) Production of interleukin-10 in the cerebrospinal fluid in aseptic meningitis of children. Pediatr Res 40:610–614
Jahnke K, Muldoon LL, Varallyay CG et al (2009) Efficacy and MRI of rituximab and methotrexate treatment in a nude rat model of CNS lymphoma. Neuro Oncol 11:503–513
Jin Kim K, Kanellopoulos-Langevin C, Merwin RM, Sachs DH, Asofsky R (1979) Establisment and characterization of BALB/c lymphoma lines with B cell properties. J Immunol 122:549–554
Johnson NA, Slack GW, Savage KJ et al (2012) Concurrent expression of MYC and BCL2 in diffuse large B-cell lymphoma treated with rituximab plus cyclophosphamide, doxorubicin, vincristine, and prednisone. J Clin Oncol 30:3452–3459
Kadoch C, Dinca EB, Voicu R et al (2009) Pathologic correlates of primary central nervous system lymphoma defined in an orthotopic xenograft model. Clin Cancer Res 15:1989–1997
Kim N, Martin TE, Simon MC, Storb U (2003) The transcription factor Spi-B is not required for somatic hypermutation. Mol Immunol 39:577–583
Klein U, Casola S, Cattoretti G et al (2006) Transcription factor IRF4 controls plasma cell differentiation and class-switch recombination. Nat Immunol 7:773–782
Kluin P, Deckert M, Ferry JA (2008) Primary diffuse large B-cell lymphoma of the CNS. In: Swerdlow SH, Campo E, Harris NL et al (eds) WHO classification of tumours of haematopoietic and lymphoid tissues. IARC, Lyon, pp 240–241
Kornelisse RF, Savelkoul HF, Mulder PH et al (1996) Interleukin-10 and soluble tumor necrosis factor receptors in cerebrospinal fluid of children with bacterial meningitis. J Infect Dis 173:1498–1502
Krueger M, Bechmann I (2010) CNS pericytes: concepts, misconceptions, and a way out. Glia 58:1–10
Krumbholz M, Theil D, Derfuss T et al (2005) BAFF is produced by astrocytes and up-regulated in multiple sclerosis lesions and primary central nervous system lymphoma. J Exp Med 201:195–200
Kurzwelly D, Glas M, Roth P et al (2010) Primary CNS lymphoma in the elderly: temozolomide therapy and MGMT status. J Neurooncol 97:389–392
Larocca LM, Capello D, Rinelli A et al (1998) The molecular and phenotypic profile of primary central nervous system lymphoma identifies distinct categories of the disease and is consistent with histogenetic derivation from germinal center-related B cells. Blood 92:1011–1019
Lenz G, Wright G, Dave SS et al (2008) Stromal gene signatures in large-B-cell lymphomas. N Engl J Med 359:2313–2323
Lo Coco F, Ye BH, Lista F et al (1994) Rearrangements of the BCL6 gene in diffuse large cell non-Hodgkin’s lymphoma. Blood 83:1757–1759
Mastroianni CM, Paoletti F, Lichtner M, D’Agostino C, Vullo V, Delia S (1997) Cerebrospinal fluid cytokines in patients with tuberculous meningitis. Clin Immunol Immunopathol 84:171–176
Mathieson BJ, Campbell PS, Potter M, Asofsky R (1978) Expression of Ly 1, Ly 2, Thy 1, and TL differentiation antigens on mouse T-cell tumors. J Exp Med 147:1267–1279
Matsumoto T, Imagama S, Hirano K et al (2012) CD44 expression in astrocytes and microglia is associated with ALS progression in a mouse model. Neurosci Lett 520:115–120
McHeyzer-Williams MG, McLean MJ, Nossal GJV, Lalor PA (1992) The dynamics of T cell-dependent B cell responses in vivo. Immunol Cell Biol 70:119–127
Mineo JF, Scheffer A, Karkoutly C et al (2008) Using human CD20-transfected murine lymphomatous B cells to evaluate the efficacy of intravitreal and intracerebral rituximab injections in mice. Invest Ophthalmol Vis Sci 49:4738–4745
Montesinos-Rongen M, Akasaka T, Zühlke-Jenisch R et al (2003) Molecular characterization of BCL6 breakpoints in primary diffuse large B-cell lymphomas of the central nervous system identifies GAPD as novel translocation partner. Brain Pathol 13:534–538
Montesinos-Rongen M, Brunn A, Bentink S et al (2008) Gene expression profiling suggests primary central nervous system lymphomas to be derived from a late germinal center B cell. Leukemia 22:400–405
Montesinos-Rongen M, Godlewska E, Brunn A, Wiestler OD, Siebert R, Deckert M (2011) Activating L265P mutations of the MYD88 gene are common in primary central nervous system lymphoma. Acta Neuropathol 122:791–792
Montesinos-Rongen M, Hans VH, Eis-Hübinger AM et al (2001) Human herpes virus-8 is not associated with primary central nervous system lymphoma in HIV-negative patients. Acta Neuropathol 102:489–495
Montesinos-Rongen M, Küppers R, Schlüter D et al (1999) Primary central nervous system lymphomas are derived from germinal-center B cells and show a preferential usage of the V4-34 gene segment. Am J Pathol 155:2077–2086
Montesinos-Rongen M, Sanchez-Ruiz M, Brunn A et al (2013) Mechanisms of intracerebral lymphoma growth delineated in a syngeneic mouse model of central nervous system lymphoma. J Neuropathol Exp Neurol 72:325–336
Montesinos-Rongen M, Schäfer E, Siebert R, Deckert M (2012) Genes regulating the B cell receptor pathway are recurrently mutated in primary central nervous system lymphoma. Acta Neuropathol 124:905–906
Montesinos-Rongen M, Schmitz R, Brunn A et al (2010) Mutations of CARD11 but not TNFAIP3 may activate the NF-kappaB pathway in primary CNS lymphoma. Acta Neuropathol 120:529–535
Montesinos-Rongen M, Schmitz R, Courts C et al (2005) Absence of immunoglobulin class switch in primary lymphomas of the central nervous system. Am J Pathol 166:1773–1779
Montesinos-Rongen M, Siebert R, Deckert M (2009) Primary lymphoma of the central nervous system: just DLBCL or not? Blood 113:7–10
Montesinos-Rongen M, Van Roost D, Schaller C, Wiestler OD, Deckert M (2004) Primary diffuse large B-cell lymphomas of the central nervous system are targeted by aberrant somatic hypermutation. Blood 103:1869–1875
Montesinos-Rongen M, Zühlke-Jenisch R, Gesk S et al (2002) Interphase cytogenetic analysis of lymphoma-associated chromosomal breakpoints in primary diffuse large B-cell lymphomas of the central nervous system. J Neuropathol Exp Neurol 61:926–933
Morse HC 3rd, Anver MR, Fredrickson TN et al (2002) Bethesda proposals for classification of lymphoid neoplasms in mice. Blood 100:246–258
Muldoon LL, Lewin SJ, Dosa E et al (2011) Imaging and therapy with rituximab anti-CD20 immunotherapy in an animal model of central nervous system lymphoma. Clin Cancer Res 17:2207–2215
Ngo VN, Young RM, Schmitz R et al (2011) Oncogenically active MYD88 mutations in human lymphoma. Nature 470:115–119
O’Connor PM, Wassermann K, Sarang M, Magrath I, Bohr VA, Kohn KW (1991) Relationship between DNA cross-links, cell cycle, and apoptosis in Burkitt’s lymphoma cell lines differing in sensitivity to nitrogen mustard. Cancer Res 51:6550–6557
Ochiai K, Maienschein-Cline M, Simonetti G et al (2013) Transcriptional regulation of germinal center B and plasma cell fates by dynamical control of IRF4. Immunity 38:918–929
Paulus W, Jellinger K (1993) Comparison of integrin adhesion molecules expressed by primary brain lymphomas and nodal lymphomas. Acta Neuropathol 86:360–364
Pels H, Montesinos-Rongen M, Schaller C et al (2005) VH gene analysis of primary CNS lymphomas. J Neurol Sci 228:143–147
Pesic M, Bartholomäus I, Kyratsous NI, Heissmeyer V, Wekerle H, Kawakami N (2013) 2-photon imaging of phagocyte-mediated T cell activation in the CNS. J Clin Invest 123:1192–1201
Ponzoni M, Berger F, Chassagne-Clement C et al (2007) Reactive perivascular T-cell infiltrate predicts survival in primary central nervous system B-cell lymphomas. Br J Haematol 138:316–323
Ramkumar HL, de Shen F, Tuo J et al (2012) IL-10-1082 SNP and IL-10 in primary CNS and vitreoretinal lymphomas. Graefes Arch Clin Exp Ophthalmol 250:1541–1548
Richter J, Ammerpohl O, Martin-Subero JI et al (2009) Array-based DNA methylation profiling of primary lymphomas of the central nervous system. BMC Cancer 9:455–462
Rickert CH, Dockhorn-Dworniczak B, Simon R, Paulus W (1999) Chromosomal imbalances in primary lymphomas of the central nervous system. Am J Pathol 155:1445–1451
Riemersma SA, Oudejans JJ, Vonk MJ et al (2005) High numbers of tumour-infiltrating activated cytotoxic T lymphocytes, and frequent loss of HLA class I and II expression, are features of aggressive B cell lymphomas of the brain and testis. J Pathol 206:328–336
Rimsza LM, Roberts RA, Miller TP et al (2004) Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood 103:4251–4258
Robertus JL, Harms G, Blokzijl T et al (2009) Specific expression of miR-17-5p and miR-127 in testicular and central nervous system diffuse large B-cell lymphoma. Mod Pathol 22:547–555
Roers A, Siewe L, Strittmatter E et al (2004) T cell-specific inactivation of the interleukin 10 gene in mice results in enhanced T cell responses but normal innate responses to lipopolysaccharide or skin irritation. J Exp Med 200:1289–1297
Rössler K, Neuchrist C, Kitz K, Scheiner O, Kraft D, Lassmann H (1992) Expression of leucocyte adhesion molecules at the human blood-brain barrier (BBB). J Neurosci Res 31:365–374
Roychowdhury S, Peng R, Baiocchi RA et al (2003) Experimental treatment of Epstein–Barr virus-associated primary central nervous system lymphoma. Cancer Res 63:965–971
Rubenstein JL, Fridlyand J, Shen A et al (2006) Gene expression and angiotropism in primary CNS lymphoma. Blood 107:3716–3723
Rubenstein JL, Wong VS, Kadoch C et al (2013) CXCL13 plus interleukin 10 is highly specific for the diagnosis of CNS lymphoma. Blood 121:4740–4748
Sagardoy A, Martinez-Ferrandis JI, Roa S et al (2013) Downregulation of FOXP1 is required during germinal center B-cell function. Blood 121:4311–4320
Schlüter D, Löhler J, Deckert M, Hof H, Schwendemann G (1991) Toxoplasma encephalitis of immunocompetent and nude mice: immunohistochemical characterisation of toxoplasma antigen, infiltrates and major histocompatibility complex gene products. J Neuroimmunol 31:185–198
Schlüter D, Oprisiu SB, Chahoud S et al (1995) Systemic immunization induces protective CD4+ and CD8+ T cell-mediated immune responses in murine Listeria monocytogenes meningoencephalitis. Eur J Immunol 25:2384–2391
Schwindt H, Akasaka T, Zühlke-Jenisch R et al (2006) Chromosomal translocations fusing the BCL6 gene to different partner loci are recurrent in primary central nervous system lymphoma and may be associated with aberrant somatic hypermutation or defective class switch recombination. J Neuropathol Exp Neurol 65:776–782
Schwindt H, Vater I, Kreuz M et al (2009) Chromosomal imbalances and partial uniparental disomies in primary central nervous system lymphoma. Leukemia 23:1875–1884
Shapiro-Shelef M, Lin KI, McHeyzer-Williams LJ, Liao J, McHeyzer-Williams MG, Calame K (2003) Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity 19:607–620
Shin T, Ahn M, Kim H et al (2005) Temporal expression of osteopontin and CD44 in rat brains with experimental cryolesions. Brain Res 1041:95–101
Soussain C, Muldoon LL, Varallyay C, Jahnke K, DePaula L, Neuwelt EA (2007) Characterization and magnetic resonance imaging of a rat model of human B-cell central nervous system lymphoma. Clin Cancer Res 13:2504–2511
Stenzel W, Dahm J, Sanchez-Ruiz M et al (2005) Regulation of the inflammatory response to Staphylococcus aureus-induced brain abscess by interleukin-10. J Neuropathol Exp Neurol 64:1046–1057
Thompsett AR, Ellison DW, Stevenson FK, Zhu D (1999) V(H) gene sequences from primary central nervous system lymphomas indicate derivation from highly mutated germinal center B cells with ongoing mutational activity. Blood 94:1738–1746
Torre D, Zeroli C, Martegani R, Speranza F (1996) Levels of interleukin-10 and tumor necrosis factor alpha in patients with bacterial meningitis. Clin Infect Dis 22:883–885
van Furth AM, Seijmonsbergen EM, Langermans JA, Groeneveld PH, de Bel CE, van Furth R (1995) High levels of interleukin 10 and tumor necrosis factor alpha in cerebrospinal fluid during the onset of bacterial meningitis. Clin Infect Dis 21:220–222
Weber T, Weber RG, Kaulich K et al (2000) Characteristic chromosomal imbalances in primary central nervous system lymphomas of the diffuse large B-cell type. Brain Pathol 10:73–84
Ye BH, Lista F, Lo Coco F et al (1993) Alterations of a zinc finger-encoding gene, BCL-6, in diffuse large-cell lymphoma. Science 262:747–750
Yuan J, Gu K, He J, Sharma S (2013) Preferential up-regulation of osteopontin in primary central nervous system lymphoma does not correlate with putative receptor CD44v6 or CD44H expression. Hum Pathol 44:606–611
Acknowledgments
The authors’ work has been supported by the Deutsche Krebshilfe (grant no.: 109471), the Wilhelm-Sander-Stiftung (2011.092.1), and the German Ministry for Education and Science(BMBF) through ICGC MMML-Seq (01KU1002A-J). We thank Katherine Dege for critical reading of the manuscript. We also acknowledge the work of all authors who could not be appropriately cited and are greatly indebted for the continuous support of the many colleagues who have provided material or clinical information over many years. This is a strong obligation for us to pursue our studies of PCNSL.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Deckert, M., Montesinos-Rongen, M., Brunn, A. et al. Systems biology of primary CNS lymphoma: from genetic aberrations to modeling in mice. Acta Neuropathol 127, 175–188 (2014). https://doi.org/10.1007/s00401-013-1202-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-013-1202-x