
Abstract
Apoptotic cell death contributes to neuronal loss in the penumbral region of brain infarction. Activated caspase-3 (ACA-3) cleaves proteins including poly(ADP-ribose) polymerase-1 (PARP-1) important in DNA repair, thus promoting apoptosis. Overactivation of PARP-1 depletes NAD+ and ATP, resulting in necrosis. These cell death phenomena have been investigated mostly in experimental animals. We studied an autopsy cohort of 13 fatal ischemic stroke cases (symptoms 15 h to 18 days) and 2 controls by immunohistochemical techniques. The number of PARP-1 immunoreactive neurons was highest in the periinfarct area. Nuclear PARP-1 correlated with increasing neuronal necrosis (P = 0.013). Cytoplasmic PARP-1 correlated with TUNEL in periinfarct and core areas (P = 0.01). Cytoplasmic cleaved PARP-1 was inversely correlated with increasing necrotic damage (P = 0.001). PAR-polymers were detected in neurons confirming enzymatic activity of PARP-1. Cytoplasmic ACA-3 correlated with death receptor Fas (r s = 0.48; P = 0.005). In conclusion, the confirmation of the same pathways of cell death than previously described in experimental animal models encourages neuroprotective treatments acting on these mediators also in human stroke.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The urge for translational brain ischemia research is derived from the clinical trials with neuroprotective regimens, which have been shown to be effective in experimental animal models but have failed in clinical trials of ischemic stroke [8, 11, 14]. The molecular mechanisms involved in the ischemic human brain might differ from the controlled conditions of the animal experiments, which are performed with relatively young animals with no comorbidities [9]. The situation is very different in an aged stroke patient with multiple underlying risk factors and concomitant acute cardiovascular or respiratory diseases at the presentation of ischemic stroke. We have previously described the dominance of neuronal apoptotic cellular damage in the periinfarct area [30]. The current study further explores the molecular mechanisms involved in pathways of neuronal death in a cohort of ischemic human brains obtained in emergency autopsies.
Apoptotic cell death is responsible only for a minority of the cells as compared to necrotic cell loss in ischemic human brain [21], but is dominant in the penumbra, the target of neuroprotective regimens [13, 17, 30]. The concomitant activation of the cell-death receptor (extrinsic) and mitochondrial (intrinsic) pathways of apoptosis has been investigated thoroughly in rats and mice after middle cerebral artery occlusion (MCAO) (for review see [29]). The role of mitochondria-dependent apoptosis in neurons has not been studied at all in ischemic adult human brains. These two pathways converge in the activation of caspase-3 (ACA-3), a down-stream terminator caspase. ACA-3 inactivates essential cellular substrates like DNA-repairing enzyme poly(ADP-ribose) polymerase-1, PARP-1, by cleaving it into 89 and 28 kDa fragments characteristic of ongoing apoptosis.
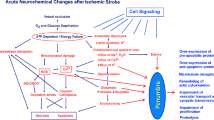
In the experimental settings, glutamate-mediated NMDA-receptor stimulation leads to increase in intracellular nitric oxide (NO) and subsequently peroxynitrate [ONOO(−)] formation, which causes DNA strand breaks and activation of PARP-1 [18, 25, 28]. Pathological overactivation of PARP-1 depletes NAD+ and ATP, and is associated with necrosis. PARP-1 cleavage is suggested to enable the ATP to be utilized for the execution of apoptosis. These hypothetical pathways leading to concomitant cell necrosis and apoptosis are illustrated in Fig. 1. In addition to these mechanisms, PAR polymers generated through PARP-1 activity are suggested to be neurotoxic [1]. Only a handful of studies have described the cell death phenomena in ischemic human brains [17, 21, 26, 30, 33], not to mention in tissues obtained with short post-mortem delay after acute stroke.

The concomitant pathways of necrotic and apoptotic cell death in brain ischemia. Illustration is modified from Meli et al. [25] with the permission from Elsevier Science. Glutamate-mediated activation of NMDA-receptor leads to increased intracellular peroxynitrate, ONOO(−), which in turn overactivates PARP-1. In a mild insult, PARP-1 exerts its physiological role in DNA repair and maintenance of chromatin integrity. In settings of massive DNA damage, PARP-1 overactivation can lead to necrotic type of cell death due to depletion of NAD+ and ATP stores. Concomitantly, overactivation of PARP-1 leads to release of AIF to the cytosol from the mitochondria. In addition, PAR is generated mainly in the nucleus, localizes to cytosol, and interacts there with mitochondria to induce cell death. Caspase-3 inhibits PARP-1 activity by cleaving it and thus “saves” energy for apoptotic machinery. AIF apoptosis inducing factor, NMDA N-methyl-d-aspartate, ONOO(−) = peroxynitrate
In experimental animal studies, neuroprotection has been suggested to ensue by inhibition of poly(ADP-ribosyl)ation [3, 18]. Furthermore, phase I and II clinical trials with PARP-1 inhibitors are underway as cardioprotecting agents or as an adjunct chemo- or radiosensitizer therapy [15, 28]. Now, clinical trials with PARP-1 inhibitors in ischemic stroke are not yet found in the databases (e.g. clinicaltrials.gov). Still, stroke is suggested to be the prime indication for development of PARP-1 inhibitors [16, 27]. This led us to explore these neuronal cell death pathways in ischemic human stroke, with the ultimate goal of aiding in the planning and interpretation of clinical trials with neuroprotectants in combination with the only present treatment of stroke, namely thrombolysis.
Materials and methods
Autopsy material
We studied autopsy specimens from 13 cases of fatal ischemic stroke (symptom duration from 15 h to 18 days) treated at the Department of Neurology, Helsinki University Central Hospital from year 1993. Autopsies were performed within a mean of 18.6 h after death (range 3.5–40 h). Two patients who died of a non-neurological cause served as controls. The study protocol was approved by the institutional review committee of the Helsinki University Central Hospital. Informed consent was given by relatives. Clinical characteristics are outlined in Table 1. Complete recanalization was absent from all cases including a patient who received 1.1 mg/kg of alteplase (rt-PA) according to the European Cooperative Acute Stroke Study (ECASS) trial protocol (Case 3).
Neuropathology
Tissue sampling was based on the most recent CT scan as described earlier [30] to obtain samples from the infarct core and periinfarct area (ipsilateral to thrombosis) and from the corresponding areas of the contralateral hemisphere and from the control brains (n = 2). On autopsy, infarction areas were identified macroscopically and approximately 1-cm3 cortical samples including subcortical white matter were dissected and fixed with formalin prior to embedding in paraffin.
Sections of 5 μm were cut and were stained with hematoxylin–eosin to confirm ischemic neuronal changes as described earlier [30]. Evaluation was performed by a neuropathologist unaware of the patient history or the sample localization. Focusing on the integrity of the nucleus, scores for signs of ischemic neuronal changes in each tissue section were given as described by [10]. In short, Score 0, normal neuronal morphology; Score 1, largely normal morphology but scattered neurons with nuclear abnormalities such as pyknosis, low nuclear cytoplasmic contrast and smearing of nuclear border (Type III neurons by Eke [10]); Score 2, large proportion of neurons with nuclear abnormalities, Score 3, large proportion of neurons with nuclear abnormalities, with scattered ones with irreversible signs such as shrunken cytoplasm with irregular borders and invisible nuclei (Type IV neurons by Eke); and Score 4, large proportion of neurons with irreversible changes.
When comparing the neuropathological scoring with the CT scan and macroscopical sampling in autopsy, the sections with scores 0 or 1 were derived from contralateral hemisphere or control areas (ipsi- or contralaterally), with scores 2–3 from periinfarct area, and with score 4 from the infarct core [30].
Immunohistochemistry
The antibodies used were as follows (antibody; dilution, manufacturer): (1) Pc rabbit anti-human Fas (C-20:sc-715); 1:400, Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA, (2) Pc rabbit anti-human FasL (Q-20:sc-956); 1:100, Santa Cruz Biotechnology Inc. (3) Rabbit immunoglobulin G (G IgG) as control for antibodies 1 and 2; 1:2,500, Vector Laboratories Inc., Burlingame, CA, USA. (4) MoAb rabbit anti-Cleaved Caspase-3 (Asp 175:5A1) (#9664); 1:75, Cell Signaling Technology, Inc., Danvers, MA, USA. Incubation with PBS as control for Ab 4. (5) MoAb anti poly(ADP-ribose) polymerase against full-length PARP-1 (MCA 1522G); 1:100, Serotec AbD, Oxford, UK. (6) Mouse immunoglobulin G1 (m IgG1) as control antibody for Ab 5; 1:100, DAKO A/S. (7) Pc rabbit anti PARP-1 p85 fragment (Cleaved PARP-1) (#G7341); 1:100, Promega Corporation, Madison, WI, USA. (8) R IgG 1:5,000 (Vector) as control for Ab 8. (9) MoAb anti-PAR (cat 4335-MC-100) 1:400, Trevigen, Gaithersburg, MD, USA. (10) M IgG3 1:400 (ID Labs, Inc., UK) as control for Ab 9. Abbreviations: Pc = polyclonal antibody (Ab). MoAb = monoclonal Ab.
Paraffin-embedded sections were deparaffinized with xylene and hydrated through graded alcohols. Antigen unmasking by microwave irradiation in 0.1 M citrate buffer (pH 6.0) twice at 750 W (5 min) and once at 650 W (5 min) was done prior to quenching in 0.3% H2O2 in methanol (3% peroxide in aqua for ACA-3 Ab) for 10 min, and blocking with 10% normal sera for 30 min (5% normal sera for 1 h for ACA-3 Ab) prior to incubation with the primary antibody at 4°C overnight. Biotinylated secondary antibody (1:200; 30 min) was followed by avidin–biotin complex for 30 min (Vectastain ABC Elite Kit; Vector Laboratories, Inc.). Incubation with diaminobenzidine (DAB) according to manufacturer’s instructions (SK-4100, Vector laboratories Inc.) was carried out prior to counterstaining with Mayer’s hemalum. For cleaved PARP-1 (p85), NovoLink™ detection was carried out according to manufacturers’ recommendation with the exception of washing buffer (0.05 M Tris–Hcl pH 7.6, 0.03 M NaCl, 0.1% Tween 20).
Additional control experiments included omission of the primary or secondary antibody in the ABC-method, which resulted in loss of immunoreactivity (not shown). For Fas antibody, see also [30]. For PARP-1 and cleaved PARP-1 antibodies, Western blot testing was performed (below). Preabsorption of 10× (volume) of caspase-3 protein (#1050, Cell Signaling Technology) with ACA-3 antibody (1 h, RT) prior to immunohistochemical staining abolished immunoreactivity in duplicate experiments (not shown).
Western blotting
Human recombinant caspase-3 protein (cat. 707-C3/CF, R&D Systems Minneapolis, MN, USA) was used to cleave purified full-length human PARP-1 protein in vitro (R&D Systems). Western blot analysis was used to assess the specificity of PARP-1 antibodies (from Serotec and Promega) in differentiating the full-length and cleaved fragment of PARP-1 (for details see Electronic supplementary material).
Double-labeling immunofluorescent staining of PARP-1 and PAR
The antibodies were applied in dilutions similar to immunohistochemistry (antibodies numbered 5–10).
Paraffin-embedded sections were deparaffinized with xylene and hydrated through graded alcohols. Antigen unmasking by microwave irradiation in 0.1 M citrate buffer (pH 6.0) once at 750 W (5 min) and twice at 650 W (5 min) was done prior to blocking with 0.1 M Tris–Hcl/3% BSA and 20% FBS, pH 7.5 for 30 min. First primary antibody was incubated for 1 h (RT) followed by rabbit anti-mouse FITC 1:20 (No. F0232, DAKO) for 1 h (RT). Endogenous Avidin/Biotin blocking was performed according to manufacturers’ recommendations (Vector cat. SP-2001) prior to the second primary antibody incubation at +4°C overnight. Anti-mouse or anti-rabbit biotinylated secondary antibody (1:200 ABC Elite Kit; Vector Laboratories, Inc.) was followed by NeutrAvidin Rhodamine Red 1:200 (cat. No A6378, Molecular probes, Netherlands) for 30 min (RT). The slides were mounted on cover slips with Vectashield-DAPI fluorescence mounting media containing 4′,6-diamino-2-phenylindole (Vector). Slides were washed twice in PBS for 5 min between all steps. All steps were performed in a humid chamber in dark.
Triple-labeling immunofluorescent staining of in situ cell death detection by TUNEL with ACA-3 and DAPI
Microwave irradiation in 0.1 M citrate buffer (pH 6.0) at 370 W (5 min) was followed by blocking with 3% bovine serum albumine (BSA) in 20% normal bovine serum/0.1 M Tris–HCl (pH 7.5) for 30 min. Overnight incubation with the primary antibody (ACA-3) was succeeded by secondary biotinylated antibody at 1:200 dilution for 30 min (Vector) and subsequent detection with RhodamineRed®-conjugated Neutralite™ avidin (A6378; Molecular Probes Europe, The Netherlands) at 1:200 dilution (30 min). Subsequently, the TUNEL reaction (terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling) was carried out according to manufacturer’s instructions (in situ cell death detection kit, Fluorescein; Boehringer Mannheim) with a FITC-tag. The sections were mounted on cover slips with Vectashield-DAPI fluorescence mounting media containing 4′,6-diamino-2-phenylindole (Vector) and then investigated with an epifluorescence microscope.
Acquisition of microscopical data and statistical analyses
The present investigational microscopical data were collected from tissue blocks from the infarct and periinfarct region and from homologous regions of the contralateral hemisphere. In each section, immunoreactivity was evaluated in five consecutive fields of 0.3125 mm2 resulting in a mean [SE] of immunopositive cell structures/mm2. We confirmed the severity of ischemic neuronal changes from an adjacent section (increasing damage scores from 0 to 4 as described in “Neuropathology” ). This also made sure that the ischemic neuronal changes associated with secondary ischemia, e.g. due to bilaterally increased intracranial pressure, were represented as early ischemic changes (i.e. score 1) in the results.
The descriptive data are given as mean [SE]. Statistical analyses were performed as described previously [30] using the SPSS for Windows-program (version 13.0). In short, the difference between the mean numbers of immunoreactive cell structures/mm2 in each brain location was evaluated by one-way ANOVA followed by LSD post hoc test. Jonckheere–Terpstra trend test was applied to determine the strength of associations between the continuously distributed variables, i.e. median immunoreactivities of ACA-3 and PARP-1 and the class-ordered ischemic neuronal score. Spearman correlation coefficient (r S) was used to assess the relation between PARP-1 and the percentage of TUNEL-positive neurons and between ACA-3 and Fas immunoreactive cells. In the box plots, the median line is shown, the bar represents the upper 75% and lower 25% quartile, and the error bar depicts high- and low values. P values < 0.05 were considered statistically significant.
Results
Our previous study described apoptotic cell morphology and TUNEL-labeling most prominently as neuronal phenomena [30], which is in line with results in experimental animal model of permanent middle cerebral artery occlusion (PMCAO) [4]. In addition, there is also experimental evidence for apoptotic cell death of other cell types, including astrocytes [31]. In the present study, we concentrated the systematic evaluation of the immunoreactivities on neuronal expression of cell death mediators, namely caspase-3 and PARP-1.
Western blotting
In Western blotting with purified PARP-1 protein, the PARP-1 antibody against full-length PARP-1 (Serotec) detected both a band of 116 kDa (equivalent to full-length PARP-1) and a less intense band of the size 89 kDa. PARP-1 protein cleavage by ACA-3 in vitro prior to Western blotting resulted in complete loss of signal for full-length PARP-1. Antibody against cleaved PARP-1 (Promega) detected only a band corresponding to cleaved PARP-1 (89 kDa) (Electronic supplementary material).
Later in the text, PARP-1 refers to full-length PARP-1 (not totally excluding minor detection of cleaved PARP-1 p89 fragment) and cleaved PARP-1 to detection of p85 fragment using the specific antibody (Promega) in immunohistochemistry.
PARP-1 immunoreactivity
Only scattered neurons showed PARP-1 immunoreactivity in control brains (n = 2; Fig. 2a). Post-ischemic PARP-1 immunoreactivity was evident both in neuronal nuclei (Fig. 2b) and in cytoplasm (Fig. 2c). Adjacent sections with m IgG1 were negative (not shown). Interestingly, post-ischemic PARP-1 immunoreactivity was also evident in the nucleolus (Fig. 2d). The maximum number of PARP-1 positive neuronal nuclei was evident in the infarct core during the first post-ischemic day and this shifted to the periinfarct area during the second day (highest at 23 h vs. at 39 h, respectively). The mean number of PARP-1 immunoreactive neuronal nuclei was highest in the periinfarct area (Fig. 3a). Interestingly, the nuclear PARP-1 positivity correlated with increasing neuronal ischemic (necrosis) score with the exception of most dense ischemic damage (Score 4; only 2 brain sections) (Fig. 3b; P = 0.013). The induction was evident up to 2 weeks (Fig. 3c).

Subcellular localization of post-ischemic PARP-1 in neurons. a Only low-level, scattered PARP-1 immunoreactivity was seen in control brains. b Neurons in the infarcted brain cortex are PARP-1 positive. Note the intense immunoreactivity in the neuronal nuclei (DAB; brown) (arrows). c Intense PARP-1 immunoreactivity is also evident in the neuronal cytoplasm (arrows). d Note the prominent PARP-1 immunoreactivity in the nucleoli of the neurons (arrow). Magnification is 400× in all. In a–d hematoxylin counterstaining is used for nuclear visualization

Quantification of neuronal PARP-1. The number of immunoreactive cells/mm2 is given in the Y-axis. a In post-ischemic brains, the number of PARP-1 immunoreactive neuronal nuclei/mm2 is highest in the periinfarct region. b Number of PARP-1 immunoreactive nuclei increases with ischemia severity = increasing neuronal ischemic damage score with the exception of areas with the most severe damage, i.e. the necrotic infarct core (Score 4). N refers to the number of brain sections in each neuronal ischemia score group. The outliers and extreme values are denoted by symbol °. c The number of PARP-1 immunoreactive neuronal nuclei is highest by less than 3 days and it subsides during the second week. d The peak number of PARP-1 immunoreactive neuronal cytoplasm is evident by less than 3 days. Statistical analysis was performed without controls in a, c, and d due to small number (n = 2). e Cytoplasmic PARP-1 correlates with increased percentage of TUNEL-labeled cells with apoptotic morphology (as described earlier [30]) in brain areas with ischemic neuronal score 2, 3 or 4 (i.e. the periinfarct and infarct core regions) up to 5.4 days. The error bars show the mean [SE] of immunopositive cell structures/mm2 in a, c and d
The number of neurons with cytoplasmic PARP-1 immunoreactivity was highest in the periinfarct region compared to infarct core area (4.6 [0.8] vs. 2.3 [0.5]; P = 0.028). Cytoplasmic PARP-1 immunoreactivity (Fig. 3d) followed a similar temporal course with maximum induction at 3 days like nuclear PARP-1. The cytoplasmic PARP-1 immunoreactivity correlated with increased percentage of TUNEL-labeled cells (described earlier [30]) in the brain regions with neuronal ischemic scores 2, 3 or 4, i.e. the periinfarct and infarct core regions (Fig. 3e; P = 0.01) up to 5.4 days survival time. In the contralateral hemisphere, the maximum number of neuronal nuclei expressing PARP-1 was evident at 1.6 days and was reduced during the third day.
Post-ischemic cleaved PARP-1 immunoreactivity
The maximum number of neuronal nuclei immunoreactive for cleaved PARP-1 was observed between 3 days and 2 weeks (Fig. 4a) compared with the peak in less than 3 days for PARP-1 (Fig. 3c). Adjacent sections with R IgG were negative (Data not shown). The number of nuclei immunoreactive for cleaved PARP-1 correlated with increasing neuronal ischemic score with the exception of most dense ischemic damage (P = 0.009; Fig. 4b, c).

Quantification of post-ischemic cleaved PARP-1 in neurons. The number of immunoreactive cells/mm2 is given in the Y-axis. a The maximum number of neuronal nuclei immunopositive for cleaved PARP-1 is evident between 3 days–2 weeks. b Nuclear cleaved PARP-1 expression correlates with increasing neuronal ischemic damage score with the exception of most dense ischemic damage. N refers to the number of sections in each neuronal ischemia score group. c Cleaved PARP-1 immunoreactivity in neuronal nuclei (arrow head) (Case 5). d The maximum number of neuronal cytoplasm immunopositive for cleaved PARP-1 is evident by less than 3 days. e Cytoplasmic cleaved PARP-1 shows an inverse correlation with increasing neuronal ischemia score. f Cleaved PARP-1 immunoreactivity in neuronal cytoplasm (arrow head) (Case 4). The outliers and extreme values are denoted by symbol ° in b and e. The bar 20 μm in c, f
The maximum number of neuronal cytoplasm expressing cleaved PARP-1 was evident by less than 3 days (Fig. 4d). The cytoplasmic cleaved PARP-1 expression was in inverse correlation with increasing neuronal ischemia score (P = 0.001; Fig. 4e, f).
PAR-polymer immunoreactivity
PAR immunoreactivity was prominent in the cytoplasm of neurons in the control brains (Fig. 5a). PAR immunoreactivity was very intense both in the neuronal nuclei and cytoplasm in two brain sections from the periinfarct area with a neuronal ischemia score 2 (Cases 1 and 6) (Fig. 5b). PAR immunoreactivity was mostly seen in the neuronal nuclei in two brain sections (Cases 12 and 13) with advanced ischemic damage (Score 4; infarct core) (Fig. 5c). The control sections with m IgG3 were negative (data not shown).

PAR immunoreactivity and double-labeling with PARP-1 and cleaved PARP-1. a PAR immunoreactivity was prominent in the cytoplasm of neurons (arrow) in a control brain (a in Table 1). b PAR immunoreactivity was very intense both in the neuronal nucleus (arrow head) and cytoplasm (arrow) in the periinfarct area with a neuronal ischemia score 2 (Case 1). c PAR immunoreactivity was mostly seen in the neuronal nuclei (arrow head) in brain sections with advanced ischemic damage (Score 4; infarct core. Case 3). d The amount of neuronal somas immunofluorescent for full-length PARP-1 was smaller than for cleaved PARP-1 (p85) (g) (Rhodamine Red; Case 2). e PAR-fluorescence in neuronal somas (FITC) colocalized (arrow head) both with the full-length PARP-1 (f) (yellow) and H, with cleaved PARP-1 enzyme (i) (double arrow heads). The bar 20 μm
Double labeling of PARP-1 and PAR
The amount of neuronal somas immunofluorescent for cleaved PARP-1 (p85) was bigger than for full-length PARP-1 (Rhodamine Red in Fig. 5d. Case 2) in keeping with the results from immunohistochemical staining (Figs. 3, 4). PAR-fluorescence in neuronal nuclei (FITC in Figs. 5e, h) colocalized both with the full-length (yellow in Fig. 5f) and cleaved PARP-1 enzyme (yellow in Fig. 5i). The control sections with m IgG3 and m IgG1 or R IgG were negative (data not shown). Less PAR-immunofluorescent cells were detected in a section from the contralateral hemisphere (Case 2) (data not shown).
Activated caspase-3 (ACA-3) immunoreactivity
Activated Caspase-3 immunoreactivity was seen only in scattered cells in the control brains (n = 2). Post-ischemic ACA-3 expression became evident in the cytoplasm of the gray matter neurons after 15 h of ICA occlusion (Fig. 6a). About a fourfold increase in the number of ACA-3 immunopositive neurons was detected in the infarct core very acutely and it subsided during the following days up to 1 week. In the periinfarct area, the magnitude of the increase in the number of ACA-3 immunoreactive neurons was roughly threefold, and it tended to be increased up to a week. A decrease in the number of ACA-3 positive neurons between the groups of acute ischemia (<3 days of symptoms) and >2 weeks ischemia duration was detected (Fig. 6b; P = 0.002). In the contralateral hemisphere, neuronal ACA-3 immunoreactivity showed an increasing trend up to 3 days, after which the number of ACA-3-positive neurons was equivalent to controls (n = 2). The cytoplasmic ACA-3-positivity correlated with Fas immunoreactivity, which we have described earlier [30] (r s = 0.48; P = 0.005; Fig. 6c).

Neuronal ACA-3 quantification and colocalization with TUNEL. The number of immunoreactive cells/mm2 is given in the Y-axis. a Neurons in infarcted cerebral cortex show cytoplasmic immunoreactivity for activated Caspase-3. Cells start to lose the normal, rounded nuclear shape with convolution of nuclear membrane (arrow head). b The number of ACA-3 immunoreactive neuronal cytoplasm is highest during 3 days and decreases during the second week. Statistical analysis was performed without controls due to small number (n = 2). c A linear correlation is evident between ACA-3 and the number of cells immunoreactive for Fas (as described earlier [30]) in infarcted brains. d In double-labeling, ACA-3 positive neuronal extensions (Rhodamine-Red; arrow head) colocalized to some extent with the TUNEL-labeled nuclei (FITC; arrow) (Case 4; periinfarct area). Magnification is 400× in a and 470× in c (bar 20 μm). In a, hematoxylin counterstaining is used for nuclear visualization
ACA-3 expression was seen in the neuronal processes in the infarct core after 1.6 days of MCA occlusion (Case 4). In double-labeling immunofluorescence, ACA-3 positive neuronal processes colocalized to some extent with the TUNEL-positive nuclei (Fig. 6d).
Discussion
Taken together, neuronal cytoplasmic ACA-3 immunoreactivity was highest in the periinfarct area—the area of most prevalent apoptosis—and it correlated with somas immunoreactive for death receptor Fas (documented previously [30]). The number of neurons with cytoplasmic PARP-1 immunoreactivity was highest in the periinfarct region and it correlated with increasing number of TUNEL-labeled neurons showing also apoptotic morphology (Fig. 3e). The number of neurons with cytoplasmic expression of cleaved PARP-1 was in inverse correlation with increasing ischemic damage (Fig. 4e). For nuclear immunoreactivity, both PARP-1 and cleaved PARP-1 correlated with increasing neuronal ischemic damage score, with a delayed peak in the expression of cleaved PARP-1 after 3 days of ischemia (Figs. 3c, 4a). As a sign of PARP-1 enzymatic activity, PAR immunoreactivity was very intense both in the neuronal somas and in the cytoplasm of post-ischemic neurons. PAR-polymers colocalized both with the full-length and cleaved PARP-1 enzyme in double-labeling (Fig. 5).
Acute neuronal injury causes overactivation of PARP-1, which results in unregulated poly(ADP-ribose) (PAR) synthesis and widespread neuronal cell death [19]. It has been hypothesized that PAR is generated mainly in the nucleus and localizes to cytosol and interacts there with mitochondria to induce cell death [1]. The present and our previous [30] observational findings generate a hypothesis of overexpressed nuclear full-length and cleaved PARP-1 as a kind of surrogate marker of necrosis while cytoplasmic PARP-1 might reflect concomitant ongoing apoptosis. After a delay of some days, PARP-1 cleavage could reflect an attempt to substitute necrosis with apoptosis in the periinfarct region, a process requiring ATP. A previous study describing PARP-1 cleavage during apoptosis in vitro demonstrated that the larger p89 PARP-1 fragment migrates from the nucleus to the cytoplasm in apoptotic cells [32]. Our results support inhibition of the pathological PARP-1 overactivation in neurons as a therapeutic strategy in human brain ischemia. It might possibly prevent cell necrosis and ensuing loss of cellular energy together with downregulation of various pro-inflammatory signal transduction pathways [34].
Only scattered neurons expressed PARP-1 in normal brain tissue, in accordance with previous results [21]. Post-ischemic nuclear expression of PARP-1 peaked in the infarct core and in the periinfarct area inside 3 days, which is consistent with previous human results [21]. On the other hand, by 2 days only very sparse cells contained detectable PARP-1 within the infarcts [21], while in our material, the induction was evident for up to 2 weeks. Regardless of the survival time, intense nuclear and cytoplasmic immunoreactivity were not detected in the same cells, supporting an ischemia-induced subcellular translocation of PARP-1. Cytoplasmic PARP-1 has been described in normal brains in a previous study, where subcellular localization of PARP-1 was studied in three non-neurological patients [5]. In another study of human traumatic brain injury, PARP-1 was found either exclusively in the nucleus or in both nuclear and cytoplasmic compartments [2]. The dominance of cytoplasmic neuronal staining was linked with a short time interval from trauma to surgery [2]. All these post-mortem studies carry a hazard that the changes in PARP immunoreactivity may be due to cell death or partial cell death, during which the nuclear content becomes more accessible to immunohistochemical staining.
Strikingly, in the present study, PARP-1 was detected in nucleoli either alone or with simultaneous nuclear expression between 15 and 39 h and only in the periinfarct and contralateral regions (Fig. 2d), which retain more physiological energy metabolism than the infarct core. Although nucleolar PARP-1 expression has not been reported in neurons before, it has been described in vitro in MDBK, HeLa and CHO cells [7] and in murine fibroblasts [23] fitting as a sign of active RNA synthesis in general [12].
The scattered expression of ACA-3 in controls is in line with the suggested roles of caspases in axon guidance, synaptic plasticity and preservation of normal brain integrity [22]. Interestingly, post-ischemic ACA-3 immunoreactivity became evident in the cytoplasm of the gray matter neurons, although the well-established role of ACA-3 as an inactivator of the nuclear protein PARP-1 (as well as other nuclear substrates) would suggest a nuclear localization [35]. In a previous study of human brain ischemia ACA-3 was strongly expressed both in the cytoplasm and in the nucleus [21]. Our results are in keeping with the results of Ferrer et al. [13] that demonstrated the exclusive expression of ACA-3 in the cytoplasm in a Western blot analysis from penumbral samples of adult rats.
Based on a series of human atherothrombotic infarcts, it has been hypothesized that apoptosis makes little contribution to neuronal death in focal brain infarcts in man, because many neurons showed obvious morphological changes of ischemic neuronal death without immunolabeling for caspase-3 in the infarcted regions [20]. In contrast to that, our results support a concomitant role for apoptosis and necrosis in neuronal ischemic cell death in keeping with our previous results demonstrating the decrease in TUNEL-labeling with increased necrosis [30]. Moreover, the cytoplasmic ACA-3-positivity correlates with post-ischemic Fas-expression—a key receptor for extrinsic pathway of apoptosis—induction of which we also have described earlier in man [30].
The major limitations of our study are that the data are descriptive (no possibility of interventions) and data are based only on immunohistochemical data. In our opinion, the autopsy brain cohort is best used for microscopical evaluation instead of homogenization for, e.g. Western Blotting. In the latter approach, the knowledge on cellular localization would be lost. Due to the extensiveness of data collected by counting the number of positive cells (semi-quantitative) under a microscope, our analysis lacks systematic evaluation of PARP-1, PAR, and ACA-3 immunoreactivities in other cells of the neurovascular unit [6, 24] besides neurons. Third, the autopsy cohort consists of serious, lethal (in many cases death due to herniation) brain infarct cases with main artery occlusion, which limits the interpretation of the results to a less severe stroke.
There is a great interest in the potential clinical benefit of PARP-1 inhibition in human diseases, such as myocardial infarction, stroke and cancer based on the double-edged role of PARP-1 in the pathogenesis of cardiovascular and inflammatory diseases and on the other hand on its ability to repair injured DNA [15]. As reviewed by Graziani and Szabo [16], a vast body of literature suggests that cells with mild, repairable DNA damage as well as cells with severely damaged DNA are diverted by PARP-1 inhibition to a common pathway, i.e. apoptotic cell elimination. PARP-1 overactivation in brain ischemia is suggested to ensue after increase of nitric oxide (NO) [25], which lead to mitochondrial dysfunction by induction of apoptosis inducing factor, AIF [24, 36] (Fig. 1). Caspase-3 (and caspase-7) mediated PARP-1 inactivation by its cleavage suggests that blocking PARP-1 activation is crucial for the execution of apoptosis. This theory of protection against necrosis by inhibition or inactivation of excessive PARP-1 renders the necrotic process amenable to pharmacological intervention [16]. Our present study demonstrates neuronal overexpression, cleavage, and subcellular translocation of PARP-1 along with neuronal overexpression of ACA-3 in human ischemic stroke.
In summary, together with our previous report [30], the present studies on a unique set of human post-mortem brains demonstrates the concomitance of necrosis and apoptosis and their relation to the extent of ischemic neuronal damage in human ischemic stroke. Moreover, the induction of several cell death mediators described originally in animal experiments (Fas, FasL, ACA-3, PARP-1) was translated to human ischemic stroke and importantly, with a prolonged window of therapeutic opportunity lasting up to several days instead of hours.
Abbreviations
- ACA:
-
Activated caspase-3
- AIF:
-
Apoptosis inducing factor
- PARP-1:
-
Poly(ADP-Ribose) polymerase-1
- TUNEL:
-
Terminal deoxynucleotidyl transferase (TdT)-mediated dUTP nick end labeling
References
Andrabi SA, Kim NS, Yu S et al (2006) Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci USA 103:18308–18318
Ang BT, Yap E, Lim J et al (2003) Poly(adenosine diphosphate-ribose) polymerase expression in human traumatic brain injury. J Neurosurg 99:125–130
Chiarugi A (2005) Poly(ADP-ribosyl)ation and stroke. Pharmacol Res 52:15–24
Chu X, Fu X, Zou L et al (2007) Oncosis, the possible cell death pathway in astrocytes after focal cerebral ischemia. Brain Res 1149:157–164
Cookson MR, Ince PG, Usher PA, Shaw PJ (1999) Poly(ADP-ribose) polymerase is found in both the nucleus and cytoplasm of human CNS neurons. Brain Res 834(1–2):182–185
del Zoppo GJ (2006) Stroke and neurovascular protection. N Engl J Med 354(6):553–555
Desnoyers S, Kaufmann SH, Poirier GG (1996) Alteration of the nucleolar localization of poly(ADP-ribose) polymerase upon treatment with transcription inhibitors. Exp Cell Res 227:146–153
Donnan GA (2008) The 2007 Feinberg lecture. A new road map for neuroprotection. Stroke 39:242–248
Durukan A, Tatlisumak T (2007) Acute ischemic stroke: overview of major experimental rodent models, pathophysiology, and therapy of focal cerebral ischemia. Pharmacol Biochem Behav 87:179–197
Eke A, Conger KA, Anderson M, Garcia JH (1990) Histologic assessment of neurons in rat models of cerebral ischemia. Stroke 21:299–304
Faden AI, Stoica B (2007) Neuroprotection: challenges and opportunities. Arch Neurol 64(6):794–800
Fakan S, Leduc Y, Lamarre D, Brunet G, Poirier GG (1988) Immunoelectron microscopical distribution of poly(ADP-ribose)polymerase in the mammalian cell nucleus. Exp Cell Res 179(2):517–526
Ferrer I, Planas AM (2003) Signaling of cell death and cell survival following focal cerebral ischemia: life and death struggle in the penumbra. J Neuropathol Exp Neurol 62(4):329–339
Fisher M, Davalos A, Rogalewski A, Schneider A, Ringelstein EB, Schäbitz W (2006) Toward a multimodal neuroprotective treatment of stroke. Stroke 37:1129–1136
Graziani G, Battaini F, Zhang J (2005) PARP-1 inhibition to treat cancer, ischemia, inflammation. Pharmacol Res 52:1–4
Graziani G, Szabo C (2005) Clinical perspectives of PARP inhibitors. Pharmacol Res 52:109–118
Guglielmo MA, Chan PT, Cortez S et al (1998) The temporal profile and morphological features of neuronal death in human stroke resemble those observed in experimental forebrain ischemia: the potential role of apoptosis. Neurol Res 20(4):283–296
Ha HC, Snyder SH (2000) Poly(ADP-ribose) Polymerase-1 in the nervous system. Neurobiol Dis 7:225–239
Koh DW, Dawson TM, Dawson VL (2005) Mediation of cell death by poly(ADP-ribose) polymerase-1. Pharmacol Res 52(1):5–14
Love S, Barber R, Srinivasan A, Wilcock GK (2000) Activation of caspase-3 in permanent and transient brain ischemia in man. NeuroReport 11:2495–2499
Love S, Barber R, Wilcock GK (2000) Neuronal death in brain infarcts in man. Neuropath Appl Neurobiol 26:55–66
McLaughlin B (2004) The kinder side of killer proteases: caspase activation contributes to neuroprotection and CNS remodeling. Apoptosis 9(2):111–121
Meder VS, Boeglin M, de Murcia G, Schreiber V (2005) PARP-1 and PARP-2 interact with nucleophosmin/B23 and accumulate in transcriptionally active nucleoli. J Cell Sci 118(1):211–222
Mehta SL, Manhas N, Raghubir R (2007) Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev 54:34–66
Meli E, Pangallo M, Baronti R et al (2003) Poly(ADP-ribose) polymerase as a key player in excitotoxicity and post-ischemic brain damage. Toxicol Lett 139:153–162
Mitsios N, Gaffney J, Krupinski J et al (2007) Expression of signaling molecules associated with apoptosis in human stroke tissue. Cell Biochem Biophys 47(1):73–86
Moroni F (2007) Poly(ADP-ribose)polymerase 1 (PARP-1) and postischemic brain damage. Curr Opin Pharmacol 7:1–8
Pacher P, Szabo C (2008) Role of the peroxynitrite-poly(ADP-Ribose) polymerase pathway in human disease. Am J Pathol 173(1):2–13
Rami A, Bechmann I, Stehle JH (2008) Exploiting endogenous anti-apoptotic proteins for novel therapeutic strategies in cerebral ischemia. Prog Neurobiol 85(3):273–296
Sairanen T, Karjalainen-Lindsberg M, Paetau A, Ijäs P, Lindsberg PJ (2006) Apoptosis dominant in the periinfarct area of human ischemic stroke—a possible target of antiapoptotic treatments. Brain 129:189–199
Solaroglu I, Tsubokawa T, Cahill J, Zhang JH (2006) Anti-apoptotic effect of granulocyte-colony stimulating factor after focal cerebral ischemia in the rat. Neuroscience 143(4):965–974
Soldani C, Lazzè MC, Bottone MG et al (2001) Poly(ADP-ribose) polymerase cleavage during apoptosis: when and where? Exp Cell Res 269(2):193–201
Stadelmann D, Mews I, Srinivasan A, Deckwerth TL, Lassmann H, Bruck W (2001) Expression of cell death-associated proteins in neuronal apoptosis associated with pontosubicular neuron necrosis. Brain Pathol 11(3):273–281
Szabo C, Pacher P, Swanson RA (2006) Novel modulators of poly(ADP-ribose)polymerase. Trends Pharmacol Sci 27:626–630
Velier JJ, Ellison JA, Kikly KK, Spera PA, Barone FC, Feuerstein GZ (1999) Caspase-8 and caspase-3 are expressed by different populations of cortical neurons undergoing delayed cell death after focal stroke in the rat. J Neurosci 19(14):5932–5941
Yu S, Wang H, Poitras MF et al (2002) Mediation of Poly(ADP-Ribose) Polymerase-1-dependent cell death by apoptosis-inducing factor. Science 297
Acknowledgments
The study was financially supported by The Finnish Academy, Sigrid Jusélius Foundation, HUCH governmental subsidiary grants for research (EVO), Päivikki and Sakari Sohlberg Foundation, H. Lundbeck Inc., Maire Taponen Foundation, The Finnish Medical Foundation, Finnish Cultural Foundation and Aarne Koskelo Foundation. The authors thank Ms. Tanja Eriksson and Mrs. Taru Puhakka for skillful technical assistance. This study belongs to a series of post-mortem stroke studies designated collectively as the Helsinki Stroke Study (HSS).
Conflict of interest statement
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
T. Sairanen and R. Szepesi contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Sairanen, T., Szepesi, R., Karjalainen-Lindsberg, ML. et al. Neuronal caspase-3 and PARP-1 correlate differentially with apoptosis and necrosis in ischemic human stroke. Acta Neuropathol 118, 541–552 (2009). https://doi.org/10.1007/s00401-009-0559-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-009-0559-3