
Abstract
Most cases of Alzheimer’s disease (AD) are sporadic in nature, although rarer familial AD (FAD) cases have provided important insights into major pathological disease mechanisms. Mutations in the presenilin 1 gene (PS1) are responsible for the majority of FAD cases, causing an earlier age of onset and more rapid progression to end-stage disease than seen in sporadic AD. We have investigated the cytoskeletal alterations in neuritic AD pathology in a cohort of FAD cases in comparison to sporadic AD and pathologically aged cases. Tau-immunoreactive neurofibrillary tangle (NFT) loads were similar between PS1 FAD and sporadic AD cases. Similarly, plaque loads, both β-amyloid (Aβ) and thioflavine S, in PS1 FAD and sporadic AD cases were not significantly different; however, in pathologically aged cases, they were significantly lower than those in PS1 cases, but were not different from sporadic AD cases. The ‘cotton wool’ plaque characteristic of PS1 cases did not demonstrate a high density of dystrophic neurites compared to other Aβ plaque types, but did demonstrate a localised mass effect on the neuropil. Despite minimal differences in plaque and NFT loads, immunolabelling demonstrated clear phenotypic differences in the NFTs and dystrophic neurites in PS1 FAD cases. Presenilin-1 cases exhibited significantly (P < 0.05) more tau-positive NFTs that were immunolabelled by the antibody SMI312 (anti-phosphorylated NF protein and phosphorylated tau) than sporadic AD cases. Presenilin-1 cases also exhibited numerous ring-like NF-positive and elongated tau-labelled dystrophic neurites, whereas these dystrophic neurite types were only abundant at the very early (pathologically aged cases) or very late stages of sporadic AD progression, respectively. These differences in cytoskeletal pathology in PS1 cases suggest an accelerated rate of neuritic pathology development, potentially due to mutant PS1 influencing multiple pathogenic pathways.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia, and is pathologically characterised by β-amyloid (Aβ) plaques, dystrophic neurites, neurofibrillary tangles (NFTs) and neuropil threads. Whilst the majority of AD cases are sporadic, genetic forms account for approximately 1% of AD cases [8]. Familial AD (FAD) has been linked to mutations in the amyloid precursor protein (APP), presenilin 1 (PS1) and presenilin 2 genes (as reviewed in [11]). Mutations in the PS1 gene cause the vast majority of FAD cases, with over 165 PS1 gene mutations detected thus far (reviewed in [26]).
In PS1 FAD, abnormal γ-secretase cleavage of APP occurs, and the resulting increase in Aβ production is likely to be central to the early onset and rapid progression to end-stage disease [5, 7, 22]. In comparison, sporadic AD may begin with an increase in Aβ production or a decrease in its clearance [43], similarly resulting in the aggregation of Aβ into plaques. β-amyloid plaques are differentially characterised into various morphotypes as defined by the three-dimensional structure and density of Aβ fibrils to form diffuse, fibrillar or dense cored plaques [2, 14]. In addition to the early onset and swift progression to end stage disease, PS1 FAD cases have been reported to demonstrate a significantly higher plaque load in comparison to sporadic AD and exhibit two plaque types that are rarely observed in sporadic cases, namely ‘inflammatory’ and ‘cotton wool’ plaques [10, 21, 23, 25–27, 31, 44, 46]. ‘Cotton wool’ Aβ plaques are large (>100 μm) [20], rounded, diffuse plaques found in the cortex of PS1 FAD cases with fine neurites containing hyperphosphorylated tau [44, 46] and globular dystrophic neurites consisting of nonhyperphosphorylated tau [44, 49], whereas ‘inflammatory’ Aβ plaques have dense cresyl violet-, silver- and thioflavine S-stained cores that are not immunoreactive for Aβ or tau but are associated with reactive microglia and astrocytes [44]. Despite these differences, genetic and sporadic AD cases may still share a common pathophysiological pathway [29].
Dystrophic neurites are abnormal neuronal processes with aberrant accumulations of cytoskeletal proteins including neurofilament (NF) triplet proteins (light, medium and heavy), α-internexin and the microtubule-associated protein, tau, which are specifically associated with dense-core, fibrillar and, to a lesser extent, diffuse Aβ plaques in sporadic AD cases [13, 15, 19, 47]. In sporadic AD cases, the morphological and biochemical properties of dystrophic neurites reflect the progression of the disease [13, 15, 32, 47, 48]. A subset of nondemented, aged cases exhibit widespread neocortical Aβ plaques, with no overt nerve cell degeneration or ‘classical’ neurofibrillary pathology, and correspond to Braak stage III [6, 39, 51, 53]. Such pathologically aged cases may represent a preclinical stage of sporadic AD. In these cases, NF triplet protein- and α-internexin-labelled dystrophic neurites with bulb- and ring-like morphology are abundant, but dystrophic neurites containing abnormal tau protein are rare [13, 15, 32, 47, 48, 54]. In contrast, sporadic AD cases demonstrate NF triplet protein- and α-internexin-labelled bulb-like dystrophic neurites, α-internexin-immunoreactive ring-like dystrophic neurites and numerous classical angular elongated tau-labelled dystrophic neurites [13, 15]. Thus, it has been proposed that dystrophic neurites represent aberrant sprouting of axons that mature through early NF abundant forms to the degenerative processes containing altered tau and paired helical filaments [51]. The current study was carried out to characterise the cytoskeletal changes that occur in the neuritic pathology in PS1 FAD compared to sporadic AD and pathological control cases, specifically NF triplet proteins and α-internexin, as such an analysis may provide further insights into the pathological progression of FAD.
Materials and methods
Tissue source and processing
Human brain tissue was acquired from the Prince of Wales Medical Research Institute Tissue Resource Centre (Sydney, Australia), the South Australian Brain Bank (Adelaide, Australia), and the Sun Health Research Institute (AZ, USA), as previously described [15, 44]. Consent for autopsy and approval for the dispersement and use of human tissue specimens for research purposes was approved by appropriate institutional Human Ethics Committees, and the research project was approved by the University of Tasmania Human Ethics Committee. Cases include eight PS1 FAD cases (average age 51.5 years, age range 44–61 years; average postmortem interval 16.4 h, postmortem interval range 5–25 h), five sporadic AD cases (average age 78.8 years, age range 71–84 years; 8.8, 5.0–16.5 hours), five sporadic pathologically aged cases (average age 84.8, age range 78–91 years; average postmortem interval 2.7 h, postmortem interval range 2.2–3.0 h) and two aged healthy control cases (79 and 83 years old, postmortem intervals of 60 and 24 h, respectively) that exhibited no AD pathology (Table 1). All AD cases exhibited Braak Stage V or VI pathology and conformed to the CERAD criteria for the diagnosis of AD [6, 35]. Sporadic pathologically aged cases did not conform to the CERAD criteria, but demonstrate Braak Stage III pathology, which includes widespread non-neuritic (as determined by thioflavine S staining and tau immunolabelling) Aβ plaques in the neocortex, and neurofibrillary pathology in the hippocampus and entorhinal formation [6]. When analysed by a one-way ANOVA with a Bonferroni posthoc test, the age of the PS1 FAD cases were significantly lower than the other case types (P < 0.05), and the postmortem interval of the PS1 FAD cases was significantly higher than that of the pathologically aged cases, but not the sporadic AD cases (P < 0.05). There were no significant differences (P < 0.05) between either the ages or postmortem intervals of the sporadic AD cases compared to the pathologically aged cases.
Immunohistochemistry and analysis
Blocks of the inferior temporal cortex (ITG) were immersion-fixed in 4% paraformaldehyde, 10% formalin or 2% picric acid (Table 1), cryoprotected, and 40 μm sections were collected on a cryostat for immunolabelling. All antibodies have been previously characterised by Western blot analysis (see Table 2). Optimal concentrations were determined for each antibody. Immunolabelling outcomes did not vary according to fixation methods, and control experiments in which the primary antibodies were omitted eliminated all immunoreactivity. All PS1 FAD, sporadic AD and pathologically aged cases were double immunolabelled with species-appropriate combinations of antibodies to Aβ [either anti-NCL-β-amyloid (mouse monoclonal) or anti-pan-β-amyloid (rabbit polyclonal)] with antibodies to phosphorylated NF and phosphorylated tau (SMI312), NF triplet protein of the medium subunit (NFM), α-internexin, hyperphosphorylated-tau (AT8) and phosphorylation-independent tau (pan-tau) (Table 2). Two PS1 cases (P264L, S168L) were not successfully labelled for α-internexin and so could not be included in this analysis. All primary antibodies were visualised with AlexaFluor goat anti-mouse and anti-rabbit secondary antibodies (dilution 1:500, Molecular Probes, Eugene, OR). For Aβ- and thioflavine S-plaque detection, tissue sections from all cases were pretreated with formic acid to maximise detection and either stained with thioflavine S or immunolabelled with antibodies to pan-Aβ (Table 2), as previously described [15]. To quantify the NFT loads and determine the percentage of specific NFT immunolabelling patterns, sporadic AD and PS1 cases were immunolabelled with the antibodies pan-tau (anti-phosphorylation-independent tau) and SMI312 (anti-phosphorylated NFs and phosphorylated tau) (Table 2) as previously described [15]. All tissue sections were viewed on a Leica DMLB2 microscope with images captured using an Optronics Magnafire-cooled CCD camera. Images for figures were prepared using Adobe Photoshop (version 9.0). Neocortical Aβ plaques were examined for the presence of NF- and α-internexin-labelled bulb-like dystrophic neurites (bulbar neurite swellings) and ring-like dystrophic neurites (spherical structures with an unlabelled core) as well as elongated and angular tau-labelled dystrophic neurites [15]. Cotton wool plaques were defined by their size (>100 μm in diameter), clear margins and relative lack of neuritic infiltrate [44].
Images of five randomly selected strips of ITG neocortex from the pia to the white matter were collected for each case to quantify the number of NFTs and the Aβ- and thioflavine S-plaque loads, using our previously described methods [44]. The strips of neocortex were approximately 1,300 μm wide for plaque load analysis and approximately 70 μm wide for NFT load analysis. The percentage area corresponding to plaque deposition was then calculated using NIH ImageJ (version 1.34s) software, and all NFTs present within the sampled regions were counted. As thioflavine S stains fibrillar aggregates within plaques, whereas the anti-Aβ antibody labels both nonfibrillar and fibrillar Aβ, both Aβ and thioflavine S plaque loads were assessed. In addition, 100 pan-tau-labelled NFTs were assessed in each case for immunolabelling for SMI312 in nonoverlapping vertical strips of neocortex from the pia to the white matter in all sporadic AD and PS1 cases. All quantitative data was averaged for each case type (standard errors reported in the text) and was analysed using a two-tailed Student’s t test or a one-way ANOVA with a Bonferroni posthoc test, with a P value of <0.05 being considered statistically significant.
Protein extraction and Western blot analysis
Protein extraction was carried out as previously described [45] from 500 mg of cortical grey matter from two PS1 FAD cases, two sporadic AD cases and two aged healthy control cases (Table 1). Briefly, tissue was homogenised in 1.5 ml of extraction buffer [0.75 M NaCl, 100 mM 2-(N-morpholino)ethanesulfonic acid, 1 mM EGTA and 0.5 mM MgSO4, 2 mM dithriothreitol at pH 6.8, supplemented with protease inhibitors], and the homogenates were centrifuged at 100,000g for 60 min at 4°C. The pellets were extracted twice in 10 ml of extraction buffer (10 mM Tris, 10% sucrose, 0.85 M NaCl, 1 mM EGTA, pH 7.4) followed by centrifugation at 15,000g for 20 min at 4°C. Supernatants were made up to 1% sarkosyl and placed on a shaker for 1 h prior to centrifugation at 100,000g for 30 min at room temperature. Final pellets were resuspended in 50 mM Tris (pH 7.4, 0.2 ml/g of starting tissue) and quantified by a BCA protein assay kit (Pierce) for subsequent Western blot analysis.
Approximately 40 μg of sarkosyl insoluble protein was separated on a 7.5% polyacrylamide gel along with a molecular weight marker (SeeBlue® Plus2 Pre-stained Standard; Invitrogen). Separated protein was then transferred onto nitrocellulose membrane (BioRad, 100 V, 60 min). Membranes were blocked (5% skim milk powder for 24 h) followed by incubation with an antiphosphorylated NF antibody (SMI312, Table 2) (1:4,000 in 2% skim milk, 16 h). Membranes were then washed (three times in Tris-buffered saline Tween-20 and once in Tris-buffered saline), prior to secondary antibody incubation (BioRad 1:6,000, 1.5 h). Enhanced chemiluminesence (Perkin Elmer; NEL100001EA) was used to visualise protein bands. Blots were then stripped (62.5 mM Tris–HCl, 2% SDS, 100 mM 2-mercaptoethanol, pH 6.7) and reblocked (5% skim milk, 2 h) before reprobing with the antihyperphosphorylated tau antibody (AT8, Table 2) (1:500) in 2% skim milk for 16 h with protein detected using chemiluminescence. To control for protein loading, blots were again stripped, blocked and probed using an anti-β-actin antibody (Saphire Bioscience; 1:10,000, data not shown).
Results
Total pathology load did not differ between PS1 FAD and sporadic AD cases
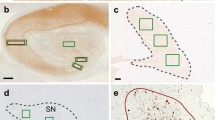
The average Aβ plaque loads in PS1 FAD, sporadic AD and pathologically aged cases were 12.1 ± 1.0, 7.0 ± 1.5 and 8.9 ± 2.1% of total area, respectively (Fig. 1a), and the average thioflavine S-stained plaque loads were 3.5 ± 0.3, 3.0 ± 0.3 and 2.1 ± 0.3% of total area, respectively (Fig. 1b). The Aβ and thioflavine S plaque loads were not significantly different between case types, with the exception of the thioflavine S plaque loads in PS1 FAD cases, which were significantly higher than pathologically aged cases (P < 0.05; Fig. 1b). Interestingly, the two PS1 cases harbouring the M146L PS1 mutation exhibited the lowest thioflavine S plaque loads in this group (Fig. 1b), and these two cases also demonstrated the lowest and third lowest Aβ plaque loads (Fig. 1a), but did not exhibit similarly low NFT loads. Cortical NFTs were not quantified in pathologically aged brains as they are rarely present in the neocortex of these cases, consistent with an absence of tau in insoluble protein extracts from control brain tissue (Fig. 2d). Cortical NFTs in PS1 FAD (17.6 ± 2.7 per 1,000 μm2) and sporadic AD (19.0 ± 2.4 per 1,000 μm2) cases were not significantly different (Fig. 1c). Although the total pathology loads in terms of plaques and NFTs were similar between all case types, the distribution of Aβ plaques differed between the sporadic AD and PS1 FAD cases. In the majority of sporadic AD cases, Aβ plaques were distributed in neocortical layers II/III and V, while the majority of PS1 FAD cases exhibited a more uniform distribution of Aβ plaques across neocortical layers I–VI (Fig. 1e, f).

Graphical representation of quantitative analysis of plaque and NFT loads in PS1 FAD, sporadic AD and pathologically aged cases. There were no significant differences in the neocortical Aβ plaque loads (% of total area) of PS1 FAD, sporadic AD and pathologically aged cases (a). Similarly, there were no significant differences in the ITG neocortical thioflavine S plaque loads (% of total area) between PS1 FAD, sporadic AD and pathologically aged cases, except for a significantly higher thioflavine S plaque load in PS1 FAD cases compared to pathologically aged cases (b). No significant difference in the number of neocortical NFTs was present between PS1 FAD and sporadic AD cases (c). The pattern of Aβ-labelled plaque deposition differed between PS1 FAD (d) and sporadic AD cases (e). The majority of PS1 FAD cases (representative image, d) demonstrated a relatively uniform distribution of Aβ plaques across all neocortical layers, while sporadic AD cases (representative image, e) exhibited Aβ plaques predominately in neocortical layers II/III and V. *P < 0.05. Scale bar 150 μm

NFT pathology in PS1 FAD cases and sporadic AD cases. Unlike sporadic AD cases (a), PS1 FAD (b) cases exhibited abundant pan-tau-labelled (red) NFTs that were also colocalised (yellow, arrowheads) with SMI312 labelling (green, a, b) and few pan-tau-labelled NFTs that did not colocalise with SMI312 immunoreactivity (arrows, a). This difference was statistically significant (c). Western blots of the insoluble protein fraction of two PS1 FAD, sporadic AD and aged control cases demonstrated that the antibody SMI312 labelled protein bands of approximately 55, 60, 69 and 78 kDa only in the protein extracts from PS1 FAD and sporadic AD cases (d). When the Western blot membrane of the insoluble protein fraction was stripped and labelled with a hyperphosphorylated tau antibody, the same protein bands were labelled at approximately 55, 60, 69 and 78 kDa in the PS1 FAD and sporadic AD cases, but not in the control extracts (e). Scale bars a 50 μm, b 30 μm
PS1 FAD cases contain more SMI312-immunolabelled NFTs than sporadic AD cases
The percentage of cortical pan-tau-labelled NFTs that also colocalised with SMI312 labelling was approximately 21 times higher (P < 0.05) in PS1 FAD cases (76.1 ± 5.0%) compared to sporadic AD cases (3.6 ± 1.5%, Fig. 2a–c). Western blot analysis of the insoluble brain protein extracts showed that SMI312-labelled 150 and 200 kDa protein bands, corresponding to both NFM and NFH, respectively, in PS1 FAD, sporadic AD and healthy aged control cases of varying postmortem intervals (range 5–60 years) (Table 1) (data not shown). However, this antibody also recognised bands of approximately 55, 60, 69 and 78 kDa in the insoluble protein extract of PS1 FAD and sporadic AD cases, but not in aged healthy controls (Fig. 2d). These bands correspond to the molecular weight of tau. To confirm this cross-reactivity, the membrane was stripped and relabelled with an antihyperphosphorylated tau antibody (Table 2), revealing the same immunoreactivity (Fig. 2e).
Assessment of dystrophic neurites as an indicator of pathological progression
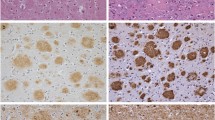
Dystrophic neurites have been classified based on their morphology and neuronal cytoskeletal immunolabelling (Table 3) into early NF immunoreactive ring-like structures, intermediate-stage α-internexin-immunolabelled ring-like structures or the intermediate filament immunoreactive bulb-like structures, or the late-stage classic tau-immunolabelled, large, elongated, tortuous neurites [13, 15, 32, 47, 48]. Neuritic plaques in pathologically aged cases exhibited abundant early and intermediate ring- and bulb-like NFM and α-internexin-labelled dystrophic neurites (Fig. 3a, d), but very rare late-stage classic tau-labelled dystrophic neurites (Fig. 3g, j; Table 3), indicating an early dystrophic neurite phenotype, which were only found in a subset of diffuse, fibrillar and dense-cored Aβ plaques (Fig. 3g, j). In contrast, sporadic AD cases demonstrated numerous intermediate α-internexin- and NFM-immunolabelled bulb-like dystrophic neurites, and ring-like dystrophic neurites that contained α-internexin but seldom NFM (Fig. 3b, e; Table 3). Sporadic AD cases also exhibited abundant late-stage, classic, large elongated tau-labelled dystrophic neurites [13, 15, 48] (Fig. 3h, k; Table 3). In comparison, the diffuse, fibrillar and dense-cored plaques present in all PS1 FAD cases were associated with numerous early- and intermediate-stage ring- and bulb-like α-internexin- and NFM-labelled dystrophic neurites (Fig. 3c, f; Table 3) as well as abundant phenotypically advanced classical elongated hyperphosphorylated-tau- and pan-tau-immunoreactive dystrophic neurites (Fig. 3i, l; Table 3).

Representative photomicrographs of double labelling immunohistochemistry for Aβ (a–l) with α-internexin (a–c), SMI312 (d–f), hyperphosphorylated tau (g–i) and pan-tau (j–l) in pathologically aged (a, d, g, j), sporadic AD (b, e, h, k) and PS1 FAD (c, f, i, l) cases. Scale bars a, b, d–f 15 μm; c, g–l 20 μm. a–c Sporadic pathologically aged cases (a), sporadic AD cases (b) and PS1 FAD cases (c) demonstrated numerous α-internexin-labelled (green) bulb- (arrows) and ring-like (arrow heads) dystrophic neurites associated with Aβ plaques (red). d–f Similarly, sporadic pathologically aged (d) and PS1 FAD cases (f) exhibited abundant SMI312-labelled (green) bulb- (arrows) and ring-like (arrow heads) dystrophic neurites, while SMI312-labelled ring-like dystrophic neurites were rare in sporadic AD cases (e). g–i A subset of Aβ plaques (red) in pathologically aged cases (g, j) were associated with tau-labelled [green; both hyperphosphorylated (g) and phosphorylation-independent epitopes (j)] punctate (arrow heads) and fine thread-like (arrows) neurites, while sporadic AD (h, k) and PS1 AD (i, l) cases exhibited abundant classical elongated tau-labelled dystrophic neurites (arrows)
Cotton wool plaques were observed in all PS1 FAD cases (Fig. 4), but not in the sporadic AD cases. A robustly lower density of NFM and α-internexin-labelled neurites were present within cotton wool plaques in comparison to the surrounding neuropil in all cases, and unlike other plaque types, the α-internexin- and NFM-labelled neurites that penetrated the cotton wool plaques were mostly of a fine calibre, and were frequently undulating and distorted (Fig. 4a–f). The early phenotypic ring-like NFM and α-internexin immunoreactive dystrophic neurites were regularly associated with cotton wool plaques, while small bulb-like NFM and α-internexin-labelled dystrophic neurites were relatively rare (Fig. 4c, d). Thickened α-internexin- and NFM-labelled neurites were also present immediately adjacent to a subset of cotton wool plaques, while other NFM and α-internexin immunoreactive neurites maintained a normal calibre but diverted their course around the edge of the plaques (Fig. 4d–f). Cotton wool plaques were associated with numerous hyperphosphorylated- and pan-tau-labelled punctate and fine thread-like neurites, and also frequently with a small number of pan-tau-labelled bulb-like dystrophic neurites and late-stage classical elongated hyperphosphorylated- and pan-tau-labelled dystrophic neurites (Fig. 4g, h). However, there was typically a higher density of punctate and fine thread-like tau-labelled neurites and fewer late-stage classical tau-labelled dystrophic neurites associated with cotton wool plaques than those associated with noncotton wool plaque types (Fig. 4i). Both PS1 FAD cases with Δexon 9 mutations exhibited a very high proportion of cotton wool plaques compared to the other familial PS1 cases.

Representative photomicrographs of cotton wool plaque pathology double labelled with antibodies against Aβ (a–i, red), NFM (a, e, green), SMI312 (b, c, f, green), α-internexin (d, green), phosphorylated tau (g, green) and pan-tau (h, i, green). Scale bars a 60 μm; b, c, e–h 30 μm; d 15 μm; i 95 μm. a The density of NFM-labelled neurites (green) was lower within Aβ-labelled (red) cotton wool plaques (*). b The SMI312-labelled (green) neurites associated with cotton wool plaques were mostly of a fine calibre and were frequently undulating and wavy (arrows). c, d SMI312- (green, c) and α-internexin-labelled (green, d) ring-like dystrophic neurites (arrow heads) were frequently associated with Aβ-labelled (red) cotton wool plaques, while bulb-like dystrophic neurites (arrows) were seldom present. e, f A subset of cotton wool plaques (red) were also associated with thickened NFM-labelled neurites (green, arrows) at their margins (e) and SMI312-labelled neurites (arrows) that were ‘bent’ around the plaque circumference (f). g, h Cotton wool plaques (red) were also associated with hyperphosphorylated-tau labelled (green) punctate (arrow heads) and fine thread-like (small arrows) neurites and also regularly with a low number of classical elongated tau-positive dystrophic neurites (large arrows, g). In addition, numerous pan-tau-labelled (green) punctate (arrow heads) and fine thread-like (small arrows) neurites along with occasional classical tau-positive dystrophic neurites (large arrows) were associated with cotton wool plaques (red, h). i Although a high density of tau-labelled abnormal neurites (green) were associated with Aβ-labelled (red) cotton wool plaques (arrow heads), there were typically fewer large classical tau-labelled dystrophic neurites and more punctate and fine thread-like tau-labelled neurites associated with cotton wool plaques, than those with the diffuse, fibrillar or dense-cored plaque types (arrows) in PS1 FAD cases
Discussion
This study aimed to assess the cytoskeletal changes associated with neuritic pathology in PS1 FAD compared to sporadic AD and pathologically aged cases. The data demonstrate differences in neurofibrillary pathology between PS1 FAD and sporadic AD despite no differences in Aβ plaque loads or absolute numbers of NFTs. Furthermore, pathological aged cases display equivalent plaque loads to AD, suggesting that plaque density may directly determine whether a subject exhibits dementia, but that the presence of neurofibrillary pathology is closely linked to this phenomenon.
In contrast to the present findings, previous studies have shown that PS1 FAD cases exhibit significantly increased Aβ deposition compared to sporadic AD brains [21, 25, 30, 44], although in most studies there is considerable group overlap. The present study demonstrated comparable NFT and plaque loads in PS1 and sporadic AD ITG, a region affected early by the disease process. Thus, the data from this region is likely to have more of a ceiling effect. The number and type of cases assessed are also likely to contribute to the differences between studies (see also [31]). Some PS1 mutations have been reported to exhibit significantly higher NFT loads compared to sporadic AD cases; however, this is not the case for all PS1 mutations examined [21]. Furthermore, the PS1 cases in the current investigation harbouring the M146L mutation exhibited lower thioflavine S plaque loads than the other PS1 cases. However, it should be noted that dramatic quantitative variations in neuropathology can exist in subjects with identical PS1 mutations, even when they are members of the same family [21, 30]. These data therefore suggest that quantitative analysis of plaque and NFT densities does not effectively discriminate between sporadic and PS1 AD cases.
The current investigation is the first to assess the degree of NF abnormalities in PS1 FAD cases and the phenotypic changes that occur in dystrophic neurites that associate with Aβ plaque deposition [13, 15, 32, 47, 48, 54]. It has been proposed that dystrophic neurites develop over time from NF- and α-internexin-labelled abnormal reactive and sprouting neurites into more degenerative forms that label for tau only (Table 3) [13, 15, 47]. In support of this, several AD mouse models exhibit NF-labelled dystrophic neurites several months before phosphorylated tau-labelled dystrophic neurites of variable morphology appear [4, 33, 36, 54]. The current data support these observations in sporadic AD and pathological aged cases and further demonstrate that PS1 FAD cases display both abundant early phenotypic ring-like NFM-immunolabelled dystrophic neurites and advanced phenotypic classical tau-labelled dystrophic neurites (Table 3), demonstrating a continuously ongoing pathogenic process in PS1 FAD relative to sporadic disease.
There was a significantly higher percentage of NFTs labelled with SMI312 in the PS1 FAD cases compared to sporadic AD cases. It has been suggested that phosphorylated NFs are only present in NFTs in the late stages of sporadic AD [42], suggesting that the progression of neurofibrillary pathology is increased in PS1 FAD. This is consistent with the very early, as well as advanced, dystrophic neurites found in the PS1 AD cases, confirming an accelerated development of neurofibrillary pathology in these cases. The Western blot data indicated that the antibody SMI312 crossreacted with tau epitopes in the insoluble protein fraction from AD brains; thus, the increased SMI312 labelling within NFTs in PS1 cases could indicate an increase in phosphorylated NF or certain tau epitopes, or both. Previously, we have reported that formic acid pretreatment exposed SMI312 immunoreactivity in tau-labelled NFTs in human AD cases [52]. Thus, it is also possible that the NFTs in PS1 FAD cases may be composed of the same components, but exhibit a different conformation of aggregated insoluble proteins compared to sporadic AD cases, resulting in an increased exposure of those epitopes recognised by the SMI312 antibody. However, irrespective of the exact epitope that the SMI312 antibody labels within NFTs, these results indicate that the molecular phenotype of NFTs in PS1 FAD cases differs from that of sporadic AD.
PS1 mutations constantly drive the overproduction of Aβ, potentially leading to pathological changes over a relatively short period of time [23, 27, 34, 41, 46]. Alternatively, increased phosphorylation of tau and subsequent tau pathology in PS1 FAD cases may be due to a loss of PS1 function [9], a gain of function of the mutant PS1 [50] and/or higher Aβ levels [7, 12, 18, 37, 44]. In support of this, mice expressing FAD-linked PS1 variants exhibited a significant increase in tau and NF phosphorylation in the spinal cord compared to mice expressing wild type PS1 [28]. However, Kwok et al. [27] reported that Δexon 8 and Δexon 9 PS1 mutations have a decreased affinity for tau and glycogen synthase kinase-3β compared to wild type PS1, suggesting that alternate or several biochemical pathways may be involved in the rapid accumulation of neurofibrillary pathology in PS1 cases. Transgenic mice with mutant tau and no NF light or heavy subunits demonstrate a dramatic decrease in the number of tau-labelled spheroids, but no decrease in the number of NFTs, suggesting that NFs may act as pathological chaperones and facilitate the rapid aggregation of tau into pathological inclusions in many neurodegenerative diseases [24]. This proposition may be relevant to a different mechanism of tau aggregation in PS1 FAD cases, as in vitro studies have shown that the expression of PS1 mutations alters NF processing resulting in inhibited neurite outgrowth [16].
The results of this investigation indicate that, in addition to other pathological features, the progression and type of AD neuritic pathology appear to differ in sporadic AD and PS1 FAD cases. Specifically, PS1 FAD cases demonstrate significantly more SMI312-immunolabelled NFTs compared to sporadic AD cases, and dystrophic neurite phenotypes consistent with both preclinical and advanced stages of AD.
References
Akiyama H, Schwab C, Kondo H et al (2002) Morphologically distinct plaque types differentially affect dendritic structure and organization in the early and late stages of Alzheimer’s disease. Acta Neuropathol 103:377–383
Armstrong RA (1998) β-Amyloid plaques: stages in life history or independent origin? Dement Geriatr Cogn Disord 9:227–238
Biernat J, Wu Y-Z, Timm T et al (2002) Protein kinase MARK/PAR-1 is required for neurite outgrowth and establishment of neuronal polarity. Mol Biol Cell 13:4013–4028
Blanchard V, Moussaoui S, Czech C et al (2003) Time sequence of maturation of dystrophic neurites associated with Aβ deposits in APP/PS1 transgenic mice. Exp Neurol 184:247–263
Borchelt DR, Thinakaran G, Eckman CB et al (1996) Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron 17:1005–1013
Braak H, Braak E (1991) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82:239–259
Busciglio J, Lorenzo A, Yeh J, Yanker BA (1995) β-Amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron 14:879–888
Campion D, Dumanchin C, Hannequin D et al (1999) Early-onset autosomal dominant Alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. Am J Hum Genet 65:664–670
Chen Q, Nakajima A, Choi SH, Xiong X, Tang YP (2008) Loss of presenilin function causes Alzheimer’s disease-like neurodegeneration in the mouse. J Neurosci Res 86:1615–1626
Crook R, Verkkoniemi A, Perez-Tur J et al (1998) A variant of Alzheimer’s disease with spastic paraparesis and unusual plaques due to the deletion of exon 9 of presenilin 1. Nat Med 4:452–455
Czech C, Tremp G, Pradier L (2000) Presenilins and Alzheimer’s disease: biological functions and pathogenic mechanisms. Prog Neurobiol 60:363–384
De Felice FG, Wu D, Lambert MP et al (2008) Alzheimer’s disease-type neuronal tau hyperphosphorylation induced by Abeta oligomers. Neurobiol Aging 29:1334–1347
Dickson TC, Chuckowree JA, Chuah MI, West AK, Vickers JC (2005) Novel Alzheimer’s disease pathology reflects variable neuronal vulnerability and demonstrates the role of β-amyloid plaques in neurodegeneration. Neurobiol Dis 18:286–295
Dickson TC, Vickers JC (2001) The morphological phenotype of beta-amyloid plaques and associated neuritic changes in Alzheimer’s disease. Neuroscience 105:99–107
Dickson TC, King CE, McCormack GH, Vickers JC (1999) Neurochemical diversity of dystrophic neurites in the early and late stages of Alzheimer’s disease. Exp Neurol 156:100–110
Dowjat WK, Wisniewski H, Wisniewski T (2001) Alzheimer’s disease presenilin-1 expression modulates the assembly of neurofilaments. Neuroscience 103:1–8
Evans J, Sumners C, Moore J et al (2002) Characterisation of mitotic neurons derived from adult rat hypothalamus and brain stem. J Neurophysiol 87:1076–1085
Ferreira A, Lu Q, Orecchio L, Kosik KS (1997) Selective phosphorylation of adult tau isoforms in mature hippocampal neurons exposed to fibrillar Abeta. Mol Cell Neurosci 9:220–234
Fukumoto H, Asami-Odaka AS, Uzuki N, Iwatsubo T (1996) Association of Aβ40-positive senile plaques with microglial cells in the brains of patients with Alzheimer’s disease and in non-demented aged individuals. Neurodegeneration 5:13–17
Giannakopoulos P, Herrmann FR, Bussiere T et al (2003) Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer’s disease. Neurology 60:1495–1500
Gomez-Isla T, Growdon WB, McNamara MJ et al (1999) The impact of different presenilin 1 and presenilin 2 mutation on amyloid deposition neurofibrillary changes and neuronal loss in familial Alzheimer’s disease brain: evidence for other phenotype-modifying factors. Brain 122:1709–1719
Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M (1998) Genetic dissection of Alzheimer’s disease and related dementias: amyloid and its relationship to tau. Nat Neurosci 1:355–358
Houlden H, Baker M, McGowan E et al (2000) Variant Alzheimer’s disease with spastic paraparesis and cotton wool plaques is caused by PS-1 mutations that lead to exceptionally high amyloid-beta concentrations. Ann Neurol 48:806–808
Ishihara T, Higuchi M, Zhang B et al (2001) Attenuated neurodegenerative disease phenotype in tau transgenic mouse lacking neurofilaments. J Neurosci 21:6026–6035
Ishii K, Ii K, Hasegawa T, Shoji S, Doi A, Mori H (1997) Increased Abeta 42(43)-plaque deposition in early-onset familial Alzheimer’s disease brains with the deletion of exon 9 and the missense point mutation (H163R) in the PS-1 gene. Neurosci Lett 228:17–20
Karlstrom H, Brooks WS, Kwok JBJ et al (2008) Variable phenotype of Alzheimer’s disease with spastic paraparesis. J Neurochem 104:573–583
Kwok JBJ, Halliday GM, Brooks WS et al (2003) Presenilin-1 mutation L271V results in altered exon 8 splicing and Alzheimer’s disease with non-cored plaques and no neuritic dystrophy. J Biol Chem 278:6748–6754
Lazarov O, Morfini GA, Pigino G et al (2007) Impairments in fast axonal transport and motor neuron deficits in transgenic mice expressing familial Alzheimer’s Disease-linked mutant presenilin 1. J Neurosci 27:7011–7020
Lippa CF, Saunders AM, Smith TW et al (1996) Familial and sporadic Alzheimer’s disease: neuropathology cannot exclude a final common pathway. Neurology 46:406–412
Mann DMA, Pickering-Brown SM, Takeuchi A, Iwatsubo T, Familial Alzheimer’s disease pathology study group (2001) Amyloid angiopathy and variability in amyloid-β deposition is determined by mutation position in presenilin-1-linked Alzheimer’s disease. Am J Pathol 158:2165–2175
Mann DMA, Takeuchi A, Sato S et al (2001) Cases of Alzheimer’s disease due to deletion of exon 9 of the presenilin-1 gene show an unusual but characteristic β-amyloid pathology known as ‘cotton wool’ plaques. Neuropathol Appl Neurobiol 27:189–196
Masliah E, Mallory M, Hansen L, Alford M, DeTeresa R, Terry R (1993) An antibody against phosphorylated neurofilaments identifies a subset of damaged association axons in Alzheimer’s disease. Am J Pathol 142:871–881
Masliah E, Sisk A, Mallory M, Games D (2001) Neurofibrillary pathology in transgenic mice overexpressing V717F β-Amyloid precursor protein. J Neuropathol Exp Neurol 60:357–368
Meyer-Luehmann M, Spires-Jones TL, Prada C et al (2008) Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nature 451:720–724
Mirra SS, Heyman A, McKeel D et al (1991) The consortium to establish a registry for Alzheimer’s disease (CERAD). Part II. Standardization of the neuropathological assessment of Alzheimer’s disease. Neurology 41:479–486
Pérez M, Asunción Morán M, Ferrer I et al (2008) Phosphorylated tau in neuritic plaques of APPsw/Tauvlw transgenic mice and Alzheimer disease. Acta Neuropathol 116:409–418
Pigino G, Pelsman A, Mori H, Busciglio J (2001) Presenilin-1 mutations reduce cytoskeletal association, deregulate neurite growth, and potentiate neuronal dystrophy and tau phosphorylation. J Neurosci 21:834–842
Porzig R, Singer D, Hoffmann R (2007) Epitope mapping of mABs AT8 and Tau5 directed against hyperphosphoylated regions of the human tau protein. Biochem Biophys Res Commun 358:644–649
Price JL, Morris JC (1999) Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol 45:358–368
Savage MJ, Iqbal M, Loh T et al (1994) Cathespin G: localization in human cerebral cortex and generation of amyloidogenic fragments from the beta-amyloid precursor protein. Neuroscience 60:607–619
Scheuner D, Eckman C, Jensen M et al (1996) Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nat Med 2:850–852
Schmidt M, Lee VM, Trojanowski JQ (1989) Analysis of epitopes shared by Hirano bodies and neurofilament proteins in normal and Alzheimer’s disease hippocampus. Lab Invest 60:513–522
Selkoe DJ (1996) (1996) Amyloid beta-protein and the genetics of Alzheimer’s disease. J Biol Chem 271:18295–18298
Shepherd CE, Gregory GC, Vickers JC et al (2004) Positional effects of presenilin-1 mutations on tau phosphorylation in cortical plaques. Neurobiol Dis 15:115–119
Stanford PM, Shepherd CE, Halliday GM et al (2003) Mutations in the tau gene that cause an increase in three repeat tau and frontotemporal dementia. Brain 126:814–826
Steiner H, Revesz T, Neumann M et al (2001) A pathogenic presenilin-1 deletion causes aberrant Aβ42 production in the absence of congophilic amyloid plaques. J Biol Chem 276:7233–7239
Su JH, Cummings BJ, Cotman CW (1996) Plaque biogenesis in brain aging and Alzheimer’s disease. I. Progressive changes in phosphorylation states of paired helical filaments and neurofilaments. Brain Res 739:79–87
Su JH, Cummings BJ, Cotman CW (1998) Plaque biogenesis in brain aging and Alzheimer’s disease. II. Progressive transformation and developmental sequence of dystrophic neurites. Acta Neuropathol 96:463–471
Takao M, Ghetti B, Hayakawa I et al (2002) A novel mutation (G217D) in the presenilin 1 gene (PSEN1) in a Japanese family: presenile dementia and Parkinsonism are associated with cotton wool plaques in the cortex and striatum. Acta Neuropathol 104:155–170
Takashima A, Murayama M, Murayama O et al (1998) Presenilin 1 associates with glycogen synthase kinase-3β and its substrate tau. Proc Natl Acad Sci USA 95:9637–9641
Vickers JC, Dickson TC, Adlard PA, Saunders HL, King CE, McCormack G (2000) The cause of neuronal degeneration in Alzheimer’s disease. Prog Neurobiol 60:139–165
Vickers JC, Riederer BM, Marugg R, Buée-Scherrer V, Buée L, Delacourtes A (1994) Alterations in neurofilament protein immunoreactivity in human hippocampal neurons related to normal aging and Alzheimer’s disease. Neuroscience 62:1–13
Woodhouse A, West AK, Chuckowree JA, Vickers JC, Dickson TC (2005) Does β-amyloid plaque formation cause structural injury to neuronal processes? Neurotox Res 7:5–15
Woodhouse A, Vickers JC, Adlard PA, Dickson TC (2007) Dystrophic neurites in TgCRND8 and Tg2576 mice mimic human pathological brain aging. Neurobiol Aging (in press). 18 October 2007 [Epub ahead of print]
Acknowledgments
Tissues were received from the Australian Brain Donor Programs Prince of Wales Medical Research Institute Tissue Resource Centre and South Australian Brain Bank, and from the Sun Health Research Institute in Arizona, USA. We wish to thank the study participants and their families who have given considerable time and eventually brain tissue to this research program, to those who participated in clinically evaluating study participants and to the staff of the neuropathology laboratory at POWMRI for tissue processing. Thanks also to Michelle Hill for her assistance with the Western Blots. This research was supported by the University of New South Wales, Gold Star Award, the Tasmanian Masonic Centenary Medical Research Foundation, National Health and Medical Research Council and the J.O. and J.R. Wicking Charitable Trust (ANZ Charitable Services). The Australian Brain Donor Programs Prince of Wales Medical Research Institute Tissue Resource Centre and South Australian Brain Bank are supported by the National Health and Medical Research Council of Australia. G.M.H. is a Principal Research Fellow and T.C.D. a Career Development Fellow of the National Health and Medical Research Council of Australia.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Woodhouse, A., Shepherd, C.E., Sokolova, A. et al. Cytoskeletal alterations differentiate presenilin-1 and sporadic Alzheimer’s disease. Acta Neuropathol 117, 19–29 (2009). https://doi.org/10.1007/s00401-008-0458-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-008-0458-z