
Abstract
To optimize treatment strategies for patients with glioblastoma, a more precise understanding of the molecular basis of this disease clearly is necessary. Therefore, numerous studies have focused on the molecular biology of glioblastoma and its linkage to clinical behavior. Here we investigated 70 glioblastomas using the array-based comparative genomic hybridization (array-CGH) with GenoSensor Array 300 to identify recurrent DNA copy number imbalances associated with patient outcomes. Univariate log-rank analysis of array-CGH data revealed 46 copy number aberrations (CNAs) associated with outcome. Among them, 26 CNAs were associated with shortened survival whereas the remaining 20 CNAs correlated with good prognosis. A hierarchical cluster analysis disclosed two genetically distinct groups of glioblastomas (1 and 2; 56 and 14 tumors, respectively). Univariate log-rank test discerned significant difference in survival between both genetic subsets while the 5-year survival rate consisted of 0 for group 1 and 63% for group 2. Multivariate analysis revealed that unfavorable genetic signature is an independent prognostic factor increasing a risk of patient death (hazard ratio, 4.38; P=0.00001). In conclusion, our current study suggests that glioblastomas can be subdivided into clinically relevant genetic subsets. Therefore, array-CGH screening of glioblastomas could provide clinically useful information and, perhaps, potentially improve the quality of treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Glioblastoma remains a challenging and hopeless chapter in neurooncology [12, 15]. Median survival of patients with glioblastoma is approximately 12 months, and only 5% of patients live for >5 years [23, 24]. The prognostic value of various parameters has been examined in patients with glioblastomas, and the only consistent prognostic variables identified were patient age and Karnofsky performance score (KPS).
To optimize treatment strategies for patients with glioblastoma, a more precise understanding of the molecular basis of this disease clearly is necessary [18, 19]. Over the past several years, a variety of numerical chromosomal abnormalities, amplifications of potent oncogenes, and deletions/mutations of tumor suppressor genes (TSGs) have been implicated in the pathogenesis of these malignancies [6, 17–19, 22, 27, 35, 43]. Nevertheless, studies regarding the clinical significance of these genetic alterations for patients with glioblastoma have reported controversial results [2]. By novel matrix-based comparative genomic hybridization (CGH) technology (or array-CGH), the status of hundreds or even thousands of genomic targets can be studied simultaneously [38]. In the present study, we investigated 70 glioblastomas using the GenoSensor Array 300 to identify recurrent DNA copy number imbalances associated with patient survival. The cases included in the analysis had a wide range of survival times to facilitate identification of genetic alterations associated with clinical outcomes.
Materials and methods
Patient population and pathological analysis
In total, 70 adult patients who were treated in the Burdenko Neurosurgical Institute from 1 January 1995 to 1 January 2004 and who had newly diagnosed, supratentorial glioblastomas were included in this study. All diagnoses were confirmed by histological assessment of a tumor sample by two independent neuropathologists according to the current World Health Organization (WHO) histologic classification [19]. Approval to link laboratory data to clinical data was obtained by the Institutional Review Board.
The 70 patients included 37 men and 33 women, and patient age was from 19 to 73 (median 51.8±12.3) years, with 34 patients (49%) younger than 50 years. All patients had undergone open surgery with either macroscopically total resection (38 patients) or subtotal tumor resection (32 patients) that was confirmed by either postoperative computed tomography (CT) (12 patients) or magnetic resonance images (MRI) (58 patients). Postoperative KPS was ≥70. All patients received postoperative radiotherapy with a total dose 56–62 cGy (mean 58 cGy) and chemotherapy with either nitrosourea compounds (14 patients) or temozolomide (56 patients). None of our patients had been treated in the settings of any prospective therapeutic trial.
The cases included in the analysis had a wide range of survival times, and five groups of patients were specially selected for these purposes: (a) 14 patients with a survival time of 3–6 months; (b) 14 patients with a survival time of 7–12 months; (c) 10 patients with a survival time of 13–18 months; (d) 16 patients with a survival time of 19–24 months (4 still alive); (e) 16 patients who lived at least 25 months (7 still alive). The 1-year survival rate for the entire cohort was 60%, the 3-year survival rate was 14%, and the 5-year survival rate was 9%.
Sample processing
Samples from each tumor were taken at the time of initial operation, immediately frozen in liquid nitrogen, and continuously stored at −80°C until used for microarray experiments. To confirm the presence of viable tumor (>90% of neoplastic cells), the cryosections of each handily dissected glioblastoma sample were stained with H&E and reviewed before DNA extraction. DNA was isolated according to a protocol that applies DNeasy tissue kit (Qiagen, Hilden, Germany). DNA concentration and quality were determined by absorption spectrophotometry, and the integrity and purity of DNA were analyzed on 1% agarose gel.
DNA labeling and hybridization of the microarrays
Approximately 100 ng of DNA from glioblastoma samples (test DNA) and normal reference human DNA were labeled by random priming reaction with Cy3-dCTP and Cy5-dCTP (Perkin Elmer, Foster City, CA, USA), respectively, according to the protocols recommended by the manufacturer (Vysis, Inc., Abbott Laboratories S.A., Downers Grove, IL, USA). Briefly, DNA was denatured with ×2.5 random priming solution at 100°C for 10 min, mixed with the nucleotide GenoSensor mix, fluorescent nucleotides and Klenow fragment, and incubated at 37°C for 2 h. After DNAse treatment, unincorporated fluorescent nucleotides were removed by ethanol precipitation.
Aliquots of labeled DNA were mixed with the hybridization buffer, denatured at 80°C for 10 min, and incubated at 37°C for 1 h to allow blocking of repetitive sequences. The hybridization mixture was then introduced on genomic DNA microarray slides. We used the commercially available GenoSensor Array 300 (Vysis, Inc) contained 287 genomic targets which were cloned from P1 and BAC libraries. The GenoSensor Array 300 includes cancer amplicons/oncogenes, TSGs, loci of gains/deletions/duplications, telomeres, and markers added to reduce gaps (for detailed information see http://www.vysis.com). The microarray slides were incubated 66–72 h at 37°C. Posthybridization process included subsequent washing in 50% formamide/2× standard saline citrate solution (three times for 10 min each at 40°C) and 1× standard saline citrate solution (four times for 5 min room temperature). The microarray slides were counterstained with DAPI solution and covered with coverslip glasses.
Analysis of the Genosensor Array 300
Microarray slides were analyzed by using the GenoSensor Reader System (Vysis, Inc) according to the instructions recommended by the manufacturer. Fluorescent signals from tumor DNA (green) and reference DNA (red) were quantitatively detected and captured by the autoexposure system, and exposure times usually varied from 0.5 s to 2.0 s. Images were analyzed with the GenoSensor Reader software, which performed a segmentation and detection of the array targets. After background subtraction, the program calculated total intensity and intensity ratio of test and reference signals for each target (three replicate spots). Normalized ratio of each target indicated the degree of gain and loss copy number when compared with the sample’s modal copy number.
We confirmed the quality of hybridization results by five control experiments using test and reference DNA from normal individuals. In all experiments, intensity ratios of hybridization signals were between 0.80 and 1.19. On the basis of the current and previous control experiments using the GenoSensor Array 300 [28, 34, 40], we determined loss, gain, and amplification of target DNA sequence copy numbers by a green/red ratio <0.8, >1.2, and >2.0, respectively.
Fluorescence in situ hybridization (FISH)
A two-color interphase fluorescence in situ hybridization (FISH) assay was performed on 5-μm-thick sections. The following commercial fluorochrome-labeled probes were applied (all produced by Vysis, Inc): centromere (CEP)7/7p12(EGFR) dual color probe set, CEP9/9p21(p16) dual color probe set, CEP10/10q23(PTEN) dual color probe set, orange-labeled 20q13(ZNF217) locus probe, and green-labeled 22q11(BCR) locus probe. Pretreatment of slides, hybridization, posthybridization processing, and signal detection were performed as described previously [21].
Statistical analysis
For categorical data, the chi-squared test, Wilcoxon–Mann–Whitney test, and Fisher’s exact test were used. Intraclass correlation analysis was used to assess the degree of associations between pairs of variables. Survival analysis was made using the Kaplan–Meier method. For pairwise comparisons of survival time distributions, the log-rank test was used. Multivariate analysis for survival was performed using the Cox proportional hazard models. Probability (P) values <0.05 were considered significant with a confidence interval (CI) at 95%. Unsupervised clustering using array-wise and uncentered hierarchical analysis with complete linkage was also performed. For this purpose, the mean ratio for each DNA target in each sample was converted to a score of 1(gain/amplification), 0 (balanced), or −1 (loss). Gain, loss, and balanced clones were represented in the final heat map as red, green, and black, respectively.
Results
Recurrent genomic imbalances in glioblastoma detected by array-CGH
Each tumor examined displayed DNA copy number aberrations (CNAs), and mean number of CNAs per tumor was 63.2±11.9 (range, 37–116). Mean number of copy number gains was 39.8±8.3 (range, 14–69) and the frequent gains are outlined in Table 1. Sixty-seven tumors (96%) harbored at least one gained clone at chromosome 7 and complete gain of 7 was found in 17 cases. Mean number of copy number losses was 39.1±6.7 (range, 22–67), and recurrent losses are outlined in Table 2. Sixty-five tumors (93%) showed at least one deleted region at chromosome 10, and complete loss of 10 was identified in 19 cases. There were no samples with completely balanced profiles for chromosomes 7 and 10 simultaneously. In addition, 41 tumors showed 81 high-level gains (Table 3) with mean number of amplifications per tumor of 1.87±1.3 (range, 1–6).
We compared the results of array-CGH and FISH analyses in all samples examined (Fig. 1), and there was a high level of concordance between data from both techniques (Table 4). We found no significant differences in mean number of CNAs between patients who were younger or older than 50 years (63.4±12.1 and 62.9±11.8, respectively). Nevertheless, we found 40 CNAs (21 gains and 19 losses), which were different for patients younger and older than 50 years (Fisher’s exact test, P <0.05; Table 5).

Array-based comparative genomic hybridization (array-CGH) (a) validation by fluorescence in situ hybridization (FISH) (b–d). Array-based cytogenetic profile of glioblastoma patient (woman; 68 years; survival time, 22 months) displays 73 copy number aberrations (gains in green and losses in red), including EGFR amplification (b; red signals and clouds), loss of CDKN2A (c; single red signals), and loss of PTEN (d; single red signals). In addition, amplification of ITGA4 locus at 2q31-q32 and deletion of SNRPN gene at 15q12 could be seen
Genomic imbalances associated with glioblastoma clinical outcome
Unsupervised hierarchical cluster analysis including all 287 DNA clones revealed two major genetic groups of glioblastomas (data not shown). Tumors from group 1 (n=25) tended to reveal an entire gain of chromosome 7 and/or complete loss of chromosome 10 and also included the cases with loss of 22q and gains at 20q. In contrast, group 2 (n=45) was composed of tumors that frequently showed gains at 12q, 15q, 19, and 22q as well as losses at 11q and 17q. There were no differences in mean CNAs between these tumor subsets (Wilcoxon–Mann–Whitney test; P=0.43). Also, average age of patients and survival times did not differ by group (Wilcoxon–Mann–Whitney test; P =0.57).
Using univariate log-rank analysis of array-CGH data according to survival times, we identified 46 CNAs significantly associated with outcome (Table 6). Among them, 26 CNAs were associated with shortened survival (unfavorable CNAs) whereas the remaining 20 imbalances correlated with good prognosis (favorable CNAs). EGFR amplification was also analyzed but showed no association with outcome.
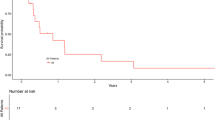
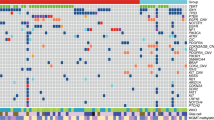
A hierarchical cluster analysis was performed repeatedly using only 46 prognostically significant CNAs. The final dendrogram shows two distinct groups of tumors (Fig. 2). All samples having no unfavorable CNAs were discerned in group 2 (n=14) whereas all tumors from group 1 (n=56) revealed these imbalances. All tumors from group 2 showed at least one unfavorable CNA in contrast to 11 glioblastomas from group 1. No differences in histopathological features could be discerned between both genetic subsets. Also, the mean number of CNAs did not differ between the clusters (Wilcoxon–Mann–Whitney test; P=0.38), but the patients from group 1 tended toward higher mean age (54.3 vs 44.7 years; Wilcoxon–Mann–Whitney test; P=0.06). There was a difference in median survival between both subsets (13.1 months for group 1 compared with 34.5 months for group 2, log-rank test, P=0.0001), and the survivors were found predominantly within group 2 (9 out of 11). Univariate log-rank test revealed significant difference in OS between both genetic subsets (P <0.00001) while the 5-year survival rate consisted of 0 for group 1 and 63% for group 2 (Fig. 3). There were no differences in survival time between the minor clusters within group 1.

Unsupervised cluster analysis of array-based comparative genomic hybridization (array-CGH) data from 70 glioblastomas (each column represents one case). We calculated cutoff assigned values of 1, 0, and −1 for gain, balanced, and loss, respectively, and clustered the samples including only 46 prognostically relevant copy number aberrations. Each row represents one clone, and clone identifiers are showed on the right. The latter includes 45 clones because HRAS locus is presented twice (as negative indicator being deleted and as positive one being gained). Losses are displayed in green, and gains are in red. The classification tree on top displays two major genetic subtypes for glioblastomas (groups 1 and 2, respectively)

Graph displaying Kaplan–Meier survival curves for glioblastoma patients in two genetic groups (1 and 2) defined by array-based comparative genomic hybridization (array-CGH) (log-rank test; P <0.0001)
Additionally, we analyzed the prognostic value of various clinical parameters. Because patient age is a continuous variable, the cutoff age that subdivided patients into distinct survival groups was calculated as described previously [20]. Consequently, the cutoff age of 50 years was the strongest predictor of outcome (5-year survival rate, 16% for younger patients compared with 2% for older patients; log-rank test; P=0.00001). We were unable to draw any significant association between overall survival and other clinical variables, including tumor location (frontal vs nonfrontal), volume of resection (total vs subtotal), and regimens of chemotherapy.
To consider whether two outlined genetic subsets of glioblastomas are independent prognostic indicators when clinical factors (age, gender, tumor location, and extent of resection) are known, we performed a multivariate regression analysis. Consequently, it was found that age >50 years (hazard ratio, 4.47; P=0.00001) and unfavorable genetic signature (hazard ratio, 4.38; P=0.00001) are independent prognostic variables increasing a risk of patient death.
Discussion
Recurrent genomic imbalances in glioblastomas
During the last several years, glioblastomas have been analyzed by using array-CGH that allowed identifying of novel aberrant loci and candidate genes involved in their tumorigenesis [5, 10, 25, 29, 32]. Our data identified numerous gains/amplifications comprising predominantly the genes involved in signal transduction pathways, cell-cycle activation, and mitosis, or code for viral oncogenes, growth factors, and receptors. Some of these genes (PDGFRA, EGFR, CDK6, MET, CDK2, SAS/CDK4, MDM2, GLI, CCND2, TOP2A, AKT1, IGFR1, PDGFB) have been previously found to be gained in glioblastomas [18, 19]. Additionally, we found frequent gains linked to oncogenes in various tumors but not previously associated with glioblastomas. The role of TP73 in cancer is complex because two isoforms originate from its different promoters, one of which is a potent oncogene [26]. FGR is a viral oncogene homolog involved in Akt signaling pathway that is frequently affected in glioblastomas [16]. MSH2 is a mismatch repair gene, but its amplification has been identified in cervical carcinoma [14]. JAG1 encodes for ligand of Notch-1 and was found to be overexpressed in human gliomas and cell lines [31]. CSF1R and FES are homologs of feline sarcoma viral oncogenes involved in the biology of various human tumors [33]. PACE4C encodes for proprotein convertase enzyme (furin) that has been associated with aggressive behavior in lung and breast cancers [4].
Our results identified frequent losses on chromosomes 9, 10, and 13, with deletions of CDKN2A, DMBT1, and RB1 described previously [18, 19]. The frequency of PTEN deletion (28%) corroborated with studies that used the GenoSensor Array 300 [28, 34, 40] but differed from data obtained by high-resolution microarrays [25]. This discrepancy may be explained by differences in the design of microarrays, number of tumors examined, and patient selection. Frequent deletions of other TSGs, such as BIN1, ROBO1, APC, TSC1, FRA16D, BRCA1, were also found in the current series (for review, see [9]). Additionally, frequently deleted clones EGR1 and EGR2 both encode for transcription factors that were defined as regulators of multiple tumor suppressors [3, 42]. Frequent loss of LPL was found in prostate carcinoma [8] whereas inactivation of SNRPN and HTR1B has been described in germ-cell and renal-cell tumors, respectively [7, 39]. Downregulated in metastases (DRIM) is a gene the decreased expression of which correlated with metastatic capability of cancers [44]. In addition, a few STS clones at 9p11.2, 17p12-p11.2, and 19p13.2 were found to be frequently deleted, thus suggesting the presence of TSGs in these regions.
In summary, the cytogenetic signature of glioblastomas displays recurrent DNA copy number imbalances with frequent gains of the clones harboring potent oncogenes and deletions of TSGs loci. These findings give evidence for a pronounced genomic instability in glioblastomas that compiles the molecular background of their biological aggressiveness.
Age-related genomic imbalances in glioblastomas
Patient age is an important prognostic factor for glioblastomas [23, 24], and age-related genetic alterations in these tumors have been established [18, 19, 43]. Consequently, we found a set of CNAs, the frequency of which differed for patients younger and older than 50 years. Gains at 7q, 19p, 19q, and 20q as well as losses at 10p and 10q tended to distribute among older patients, which is in keeping with cytogenetic studies published previously [22, 27, 43]. Loss of SNRPN was found almost exclusively in samples from older patients. This gene encodes for small ribonuclear protein, and deletion of SNRPN is critical for development of Prader-Willi syndrome. Tumors from younger patients frequently harbored deletions of TSGs (BIN1, EGR1, and FRA16D) and losses of potent oncogenes (THRB, TIF1, MYB, PIK3CA). TIF1 encodes for ligand-activated transcriptional factor. Glioblastomas from older patients usually showed gain of this locus, along with other clones on 7q, whereas tumors from their younger counterparts usually disclosed TIF1 as a minimally deleted region at 7q32–q34. Thus, our findings allow one to suggest that glioblastomas from various age groups represent molecularly distinct diseases that can develop along substantially different genetic pathways.
Prognostically relevant genetic subsets of glioblastomas
A few array-CGH glioblastoma studies included survival analysis, but the results obtained are controversial [25, 28, 29]. Our current study suggests that glioblastomas can be subdivided into clinically relevant genetic subsets. The 5-year survival in our patient cohort was greater than that reported for the entire population of glioblastomas [23, 24]. The reasons for such difference are likely related to selection bias because the cases included in this study showed a wide range of survival times. Given the strong relationship between older age and survival in glioblastomas, our cohort of patients was younger than the median age of patients with these tumors.
Current study disclosed a set of CNAs predicting for patient outcome. Most prognostically significant CNAs are not associated with well-defined chromosomal imbalances in glioblastomas. Some of these CNAs were also found to be correlated with patient age, thus explaining partly the age-related differences in outcomes of these tumors.
Unfavorable copy number gains comprised potent oncogenes (AKT3, p44S10, FGF4, FGF3, MLL, TCL1A, THRA, YES1, JUNB) that were reported to be overrepresented in solid tumors and hematological malignancies (for review, see [9]). Additionally, TP63 was found frequently amplified in squamous cell carcinomas [13]. LAMC2 and CTSB encode for laminin5 gamma2 chain and cathepsin B, respectively; both have been established as promoters of tumor invasiveness [1, 36]. Unfavorable losses targeted genes associated with cell death and suppression of tumor growth. CASP8 encodes for a key enzyme of the top of the apoptotic cascade [41]. HIC1 is a putative TSG encoding a transcription factor that has been found inactivated in various tumors [30]. LLGL1 is a human homolog of Drosophila suppressor gene, and its experimental inactivation leads to malignant transformation of primordial cells in the brain [11]. HRAS allelic deletion was found to be a common abnormality in various tumors, and these findings suggest that a putative TSG closely linked to HRAS on 11p15.5 is involved in carcinogenesis. Yokota et al. [45] found that allelic deletions of HRAS were correlated with progression and metastases of carcinomas and sarcomas.
The biologic significance of favorable CNAs remains to be defined. These imbalances comprised gains of putative TSGs (EXT1, UBE3A, DCC) and losses of potent oncogenes (TERC, MYB,PCNT2). Association of INS with glioblastoma survival has been identified [28]. Previous deletion mapping studies gradually narrowed down the 19q region involved in gliomas, and the frequent region of deletion was reported to be located at 19q13.3 [37]. However, we found here that glioblastoma prognosis was associated with loss of distal region at 19q13.4. Association of loss of the 9p21/CDKN2A locus with glioblastoma adverse outcomes has been discussed widely [6, 17–19, 21, 22, 27, 35, 43]. Surprisingly, we found that losses of subtelomere and pericentromeric regions at 9p were associated with favorable survival. From the current awareness of glioblastoma molecular biology, it is difficult to explain such associations; therefore, these data need to be elaborated further.
Combined cluster analysis allowed one to subdivide an entire tumor cohort on two genetically different subtypes with striking differences in outcomes, thus supporting the notion that glioblastomas might comprise molecularly distinct tumor entities despite their histological similarity.
Conclusions
Genomic profiling of glioblastomas using the GenoSensor Array 300 identified several genetic alterations that might be associated with biologic behavior of these malignancies. Detailed mutational analyses and functional studies will be required to elucidate the role that each of these alterations might play in glioblastoma pathogenesis and clinical course.
References
Akimoto S, Nakanishi Y, Sakamoto M, Kanai Y, Hirohashi S (2004) Laminin 5 beta and gamma 2 chains are frequently co-expressed in cancer cells. Pathol Int 54:688–692
Arslantas A, Artan S, Oner U, Muslumanoglu H, Durmaz R, Cosan E, Atasoy MA, Basaran N, Tel E (2004) The importance of genomic copy number changes in the prognosis of glioblastoma multiforme. Neurosurg Rev 27:58–64
Baron V, Adamson ED, Calogero A, Ragona G, Mercola D (2005) The transcriptional factor EGR1 is a direct regulator of multiple tumor suppressors including TGFbeta1, PTEN, p53 and fibronectin. Cancer Gene Ther 13:115–124
Bassi DE, Fu J, Lopez de Cicco R Klein-Szanto AJ (2005) Proprotein convertases: “Master switches” in the regulation of tumor growth and progression. Mol Carcinogen 44:151–161
Bredel M, Bredel C, Juric D, Harsh GR, Vogel H, Recht LD, Sikic BI (2005) High-resolution genome-wide mapping of genetic alterations in human glial brain tumors. Cancer Res 65:4088–4096
Burton E, Lamborn KR, Feuerstein BG, Prados M, Scott J, Forsyth P, Passe S, Jenkins RB, Aldape KD (2002) Genetic aberrations defined by comparative genome hybridization distinguish long-term from typical survivors in glioblastoma. Cancer Res 62:6205–6210
Bussey KJ, Lawce HJ, Himoe E, Shu XO, Heerema NA, Perlman EJ, Olson SB, Magenis RE (2001) SNRPN methylation patterns in the germ cell tumors as a reflection of primordial germ cell development. Genes Chromosome Cancer 32:342–352
Callucci M, Merola R, Farsetti A, Orlandi G, Sentinelli S, De Carli P, Leonardo C, Carlini P, Guadangi F, Sperduti I, Cianciulli AM (2005) Cytogenetic profile as an additional marker to pathological features in clinically localized prostate carcinoma. Cancer Lett (in press)
Coleman WB, Tsongalis GJ (2002) The molecular basis of human cancer. Humana Press, Totowa, 588 pp
Cowell JK, Matsui S, Wang YD, LaDuca J, Conroy J, McQuad D, Nowak NJ (2004) Application of bacterial artificial chromosome array-based comparative genomic hybridization and spectral karyotyping to the analysis of glioblastoma multiforme. Cancer Genet Cytogenet 151:36–51
Grifoni D, Garoia F, Schimanski CC, Smitz G, Laureneti E, Galle PR, Pession A, Cavicci S, Strand D (2004) The human protein Hugl-1 substitutes for Drosophila lethal giant larvae tumor suppressor function in vivo. Oncogene 23:8688–8694
Grossman SA, Batara JF (2004) Current management of glioblastoma multiforme. Semin Oncol 31:635–644
Hibi K, Trink B, Patturajan M, Westra WH, Caballero OI, Hill DE, Ratovitski EA, Jen J, Sidransky D (2000) AIS is an oncogene amplified in squamous cell carcinoma. Proc Nat Acad Sci USA 97:5462–5477
Hidalgo A, Baudis M, Pedersen I, Arreola H, Pina P, Vazquez-Ortiz G, Hernandez D, Gonzalez D, Lazos M, Lopez R, Perez C, Carcia J (2005) Microarray comparative genomic hybridization detection of chromosomal imbalances in uterine cervix carcinoma. BMC Cancer 5:77
Holland EC (2000) Glioblastoma multiforme: the terminator. Proc Nat Acad Sci USA 97:6242–6244
Hu Y, Liu Y, Pelletier S, Buchdunger E, Warmuth M, Fabbro D, Hallek M, Van Etten RA, Li S (2004) Requirement of Src kinases Lyn, Hck, and Fgr for BCR-ABL1-induced B-lymphoblastic leukemia but not chronic myeloid leukemia. Nat Genet 36:453–461
Huhn SL, Mohapatra G, Bollen A, Lamborn K, Prados MD, Feuerstein BG (1999) Chromosomal abnormalities in glioblastoma multiforme by comparative genomic hybridization: correlation with radiation treatment outcome. Clin Cancer Res 5:1435–1433
Ichimura K, Ohgaki H, Kleihues P, Collins VP (2004) Molecular pathogenesis of astrocytic tumors. J Neurooncol 70:137–160
Kleihues P, Cavenee WK (2000) Tumors of the nervous system. Pathology and genetics: World Health Organization international classification of tumours. WHO/IARC, Lyon, France
Korshunov A, Golanov A, Sycheva R, Pronin I (1999) Prognostic value of tumour-associated antigens immunoreactivity and apoptosis in cerebral glioblastomas: an analysis of 168 cases. J Clin Pathol 52:574–580
Korshunov A, Sycheva R, Golanov A (2004) Molecular stratification of diagnostically challenging high-grade gliomas composed of small cells. The utility of fluorescence in situ hybridization. Clin Cancer Res 10:7820–7826
Koschny R, Koschny T, Froster UG, Krupp W, Zuber MA (2002) Comparative genomic hybridization in glioma: a meta-analysis of 509 cases. Cancer Genet Cytogenet 135:147–159
Lamborn KR, Chang SM, Prados MD (2004) Prognostic factors for survival of patients with glioblastoma. Recursive portioning analysis. Neuro-oncology 6:227–235
Lutterbach J, Saurbrei W, Guttenberg R (2003) Multivariate analysis of prognostic factors in patients with glioblastomas. Strahlenther Onkol 179:8–15
Misra A, Pellarin M, Nigro J, Smirnov I, Moore D, Lamborn KR, Pinkel D, Albertson DG, Feuerstein BG (2005) Array comparative genomic hybridization identifies genetic subgroups in grade 4 human astrocytoma. Clin Cancer Res 11:2907–2918
Melino G, De LV, Vousden KH (2002) P73: friend or foe in tumorigenesis. Nat Rev Cancer 2:605–615
Mohapatra G, Bollen AW, Kim DH, Lamborn K, Moore DH, Prados MD, Feuerstein BG (1998) Genetic analysis of glioblastoma multiforme provides evidence for subgroups within the grade. Genes Chromosome Cancer 21:195–206
Nakahara Y, Shiraishi T, Okamoto H, Mineta T, Oishi T, Sasaki T, Tabuchi K (2004) Detrended fluctuation analysis of genome-wide copy number profiles of glioblastomas using array-based comparative genomic hybridization. Neuro-Oncology 6:281–289
Nigro J, Misra A, Zhang L, Smirnov I, Colman H, Griffin C, Ozburn N, Chen M, Pan E, Koul P, Yung WKA, Feuerstein BG, Aldape KD (2005) Integrated array-comparative genomic hybridization and expression array profiles identify clinically relevant subtypes of glioblastoma. Cancer Res 65:1678–1686
Parrella P, Scintu M, Prencipe M, Poeta ML, Gallo AP, Rabitti C, Rinaldi M, Tommasi S, Paradiso A, Schittulli F, Vallori MF, Toma S, Altomare V, Fazio VM (2005) HIC1 promoter methylation and 17p13.3 allelic loss in invasive ductal carcinoma of the breast. Cancer Lett 222:75–81
Purow BW, Haque RM, Noel MW, Su Q, Budrick MJ, Lee J, Sundaresan T, Pastorini S, Park JK, Mikolaenko I, Maric D, Eberhart CG, Fine HA (2005) Expression of Notch-1 and its ligands, Delta-like-1 and jagged-1, is critical for glioma cell survival and proliferation. Cancer Res 65:2353–2363
Roerig P, Nessling M, Radlwimmer B, Joos S, Wrobel G, Schwanen C, Reifenberger G, Lichter P (2005) Molecular classification of human gliomas using matrix-based comparative genomic hybridization. Int J Cancer 117:95–103
Roussel MF, Sherr CG (2003) Oncogenic potential of the c-FMS protooncogene (CSF-1 receptor). Cell Cycle 2:5–6
Sasaki T, Arai H, Beppu T, Ogasawara K (2003) Detection of gene amplification and deletion in high-grade gliomas using a genome DNA microarray (GenoSensor Array 300). Brain Tumor Pathol 20:59–63
Schmidt MC, Antweiler S, Urban N, Mueller W, Kuklik A, Meyer-Puttlitz B, Wiestler OD, Louis DN, Fimmers R, von Deimling A (2002) Impact of genotype and morphology on the prognosis of glioblastoma. J Neuropathol Exp Neurol 61:321–328
Sloane BF, Yan S, Podgorsky I, Linebaugh BE, Cher ML, Mai J, Cavallo-Medved D, Sameni M, Dosescu J, Moin K (2005) Cathepsin B and tumor proteolysis: contribution of tumor microenvironment. Semin Cancer Biol 15:149–157
Smith JS, Tachibana I, Pohl U, Lee HK, Thanarajasingam U, Portier BP, Ueki K, Ramaswamy S, Billings SJ, Mohrenweiser HW, Louis DN, Jenkins RB (2000) A transcript map of of the chromosome 19q-arm glioma tumor suppressor region. Genomics 64:44–50
Snidjers AM, Pinkel D, Albertson DG (2003) Current status and future prospects of array-based comparative genomic hybridization. Brief Funct Genomic Proteomic 2:37–45
Strefford JC, Stasevich I, Lane TM, Lu YJ, Oliver T, Young BD (2005) A combination of molecular cytogenetic analyses reveals complex genetic alterations in conventional renal-cell carcinoma. Cancer Genet Cytogenet 159:1–9
Suzuki T, Maruno M, Wada K, Kagawa N, Fujimoto Y, Hashimoto N, Izumoto S, Yoshimine T (2004) Genetic analysis of human glioblastoma using a genomic microarray system. Brain Tumor Pathol 21:27–34
Thorburn A (2004) Death receptor-induced cell killing. Cell Signal 16:139–144
Unoki M, Nakamura U (2001) Growth suppressive effects of BZOP and EGR2, two genes involving in the PTEN signaling pathway. Oncogene 20:4457–4465
Wiltshire RN, Herndon JE, Lloyd A, Friedman HS, Bigner DD, Bigner SH, McLendon RE (2004) Comparative genomic hybridization analysis of astrocytomas. Prognostic and diagnostic implications. J Mol Diagn 6:166–179
Xing X, Du X, Lu Z, Ning T, Su X, Ke Y (2005) Characterization of the promoter 1A6/DRIM, a novel cancer-related gene, and identification of its transcriptional activator. Gene 344:161–169
Yokota J, Tsunetsugu-Yokota Y, Battifora H (1986) Alterations of myc, myb, and ras-Ha proto-oncogenes in cancers are frequent and show clinical correlation. Science 231:261–265
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Korshunov, A., Sycheva, R. & Golanov, A. Genetically distinct and clinically relevant subtypes of glioblastoma defined by array-based comparative genomic hybridization (array-CGH). Acta Neuropathol 111, 465–474 (2006). https://doi.org/10.1007/s00401-006-0057-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-006-0057-9