
Abstract
Macrophages are one cell type in the innate immune system. Recent studies involving macrophages have overturned the conventional concept that circulating bone marrow-derived blood mononuclear cells in the adult body continuously replace macrophages residing in the tissues. Investigations using refined technologies have suggested that embryonic hematopoiesis can result in the differentiation into macrophage subgroups in some tissues. In adulthood, these macrophages are self-sustaining via in situ proliferation, with little contribution of circulating bone marrow-derived blood mononuclear cells. Macrophages are integral component of the heart, accounting for 8% of the non-cardiac cells. The use of innovative molecular techniques in paradigm shifting researches has revealed the complexity of cardiac macrophages, including their heterogeneity and ontological diversity. Resident cardiac macrophages modulate the physiological and pathophysiological processes of the cardiovascular system, with distinct and crucial roles in healthy and injured hearts. Their functions include sensing of pathogens, antigen presentation, digesting cell debris, regulating inflammatory responses, generating distinct cytokines, and secreting some regulatory factors. More recent studies have revealed further functions of cardiac macrophages. This review focuses on macrophages within the cardiovascular system. We discuss evidence that has changed our collective view of cardiac macrophage subgroups, and improved our understanding of the different phenotypes, cell surface markers, heterogeneities, origins, developments, and the dynamic and separate roles of these cardiac macrophage subgroups in the steady state and injured hearts. This review may provide novel insights concerning the pathophysiology of cardiac-resident macrophages in cardiovascular diseases and innovative therapeutic strategies that could include the modulation of the role of macrophages in cardiovascular injuries.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A statistical analysis of the 2017 Global Burden of Disease Study (https://www.healthdata.org/gbd) data revealed that non-communicable diseases (NCDs) accounted for 73.4% of global mortality. Cardiovascular mortality ranks first in global human mortality due to NCDs, with 17.8 million deaths attributed to cardiovascular diseases (CVDs) in the 2017 study [58]. The Prospective Urban Rural Epidemiology Study (PURE; https://www.phri.ca/pure/) prospectively estimated the morbidity and mortality of common diseases in people with disparate incomes from different countries. The study subdivided countries into three income-related groups (high-, middle-, and low-income). CVD morbidity and related mortality in poorer countries were higher than in developed countries. The lower income countries experienced more CVD-related deaths than deaths due to with neoplasm, with the opposite found in developed countries and several middle-income countries. Although PURE indicates a novel epidemiological shift among these various classifications of NCDs, CVDs continue to be the primary cause of global death in humans between 35 and 70 years of age [8].
The mammalian heart is comprised of cardiac and non-cardiac cells. The latter include various cell types, such as endotheliocytes, fibroblasts, smooth muscle cells, immune cells, and parenchymal cells. Endothelial cells outnumber all other non-cardiac cells, comprising greater than 60% of the cells, contrary to what was once thought. Fibroblasts constitute less than 20% of the non-cardiac cells [54]. The crosstalk communication and cooperation between these cardiac and non-cardiac cells is the basis of the origin, evolution and maturation of the heart [55].
In the 1960s, Van Furth and Cohn proposed that macrophages (MFs) are a component of mononuclear phagocytes in the body and suggested that they were derived from circulating blood mononuclear cells [66]. MFs in the heart are a part of the innate immune system with important functions in myocardium homeostasis, inflammation and pathology [48]. MFs likely degrade released macromolecules and phagocytose apoptotic cardiomyocytes, simultaneously mediating the formation of granulation tissue and structural reconstruction [50]. Several contemporary studies have reversed the long-standing view that MFs residing in the heart originate solely from circulating mononuclear cells [22]. Furthermore, clarification of the features of cardiac-resident MFs has revealed functions that go beyond the long-established functions as defenders and performers in the immune system [69]. We review here the present cognition about the heterogeneous originations of cardiac-resident MFs, its renewals and contributions to heart both during steady and after heart injury.
Classification of cardiac-resident MFs
MFs have pronounced plasticity. In the general M1/M2 subgroup classification method that has been widely studied, M1 designates “pro-inflammatory” MF and M2 designates “anti-inflammatory” MFs [68]. This classification is appropriate for some in vitro studies. However, it has numerous disadvantages when used in in vivo studies of MF subpopulations [18]. In vivo, MFs often simultaneously express surface markers of the M1/M2 subset. Seeking to develop a classified method that is better suited to in vivo conditions, some researchers have proposed a common MF nomenclature, which classifies MFs on the basis of diverse origin, activation, and binding to their cell surface biomarkers.
One study reported that cardiomyocytes make up 31.0 ± 4.2% of the total cells in the heart, with 43.6 ± 4.1% of the total cells being endotheliocytes, 4.7 ± 1.5% being leukocytes, and 20.7 ± 4.5% unidentified of. The same study reported that concerning non-cardiac cells, 63.3 ± 5.4% were endotheliocytes, 6.8 ± 2.1% were white blood cells, and 29.9 ± 5.9% were unidentified. To further determine the composition of non-cardiac cells, flow cytometry analysis was performed using different antibodies directed at endotheliocytes, white blood cells, and resident mesenchymal cells (RMC), as well as staining of nuclei and metabolic activity. These analyses revealed that endothelial cells (CD31+ CD45–) constituted 62.1 ± 3.9% of non-cardiac cells, leukocytes (CD45+) accounted for 9.6 ± 1.3%, and RMC (CD31– CD45–) accounted for 27.3 ± 5.3%. Simultaneously, data generated using the Sequential PAttern Discovery using Equivalence classes(SPADE) algorithm revealed 63.9 ± 3.4% of non-cardiac cells were endotheliocytes, 9.4 ± 1.6% were white blood cells, and the remaining 26.7 ± 4.0% were resident mesenchymal cells. To describe subgroups of white blood cells, the authors analyzed leukocytes (CD45+) on the basis of several surface biomarker molecules. Myeloid cells (CD11b+; 81.4 ± 1.4%), B cells (8.9 ± 0.6%), T cells (3.1 ± 0.4%), and non-myeloid/lymphoid cells (6.6 ± 0.6% leukocytes) were characterized within the system [54].
In another study, flow cytometry analysis established that 10.3 ± 0.4% of the cardiac cell population from mice were white blood cells, and that CD45+ CD11b+ F4/80+ Ly6Clow macrophages (7.9 ± 0.3% of all cardiac cells) were the most abundant proportion of all white blood cells in the heart. In a series of five autopsy cases, examination of human hearts using the CD68 MF biomarker revealed a spindle-like appearance of myocardial MFs [27]. Another study described that most of the CD45+white blood cells in the murine myocardium were CD11b+ F4/80+Ly6clowMFs. Analogous to those of mice, myocardial cells of humans consisted of more MFs in the atrio-ventricular node than in the left ventricular myocardium, and the circulating monocytes had a minimal contribution to MFs in these two sites. MFs in the human atrio-ventricular (AV) node were spindle shaped [30].
Similar to these studies, another study visualized MFs residing in the total leukocyte population in the normal myocardium using a murine Cx3cr1GFP/+ transgenic reporter in which the gene encoding enhanced green fluorescent protein (GFP) substituted for one allele of the Cx3cr1 gene in MFs [32]. Another study using transgenic mice found that GFP-positive cells constituted the majority of CD45+cells, and that most were myeloid, as determined using CD11b staining. These findings revealed that cardiac MFs accounted for a substantial portion of white blood cells [55].
The heterogeneity of the macrophages residing in the heart tissue has been implied based on the presence of various surface molecular biomarkers and ontogenesis. One study described at latest three different macrophage subgroups in the adult murine myocardium: CCR2– MHC-IIlow MFs, CCR2– MHC-IIhigh MFs, and monocyte-derived CCR2+MHC-IIhigh MFs. Mononuclear cells have been characterized as CCR2+ MHC-IIlow [33]. CCR2 (chemokine (C–C motif) receptor 2) has been closely related to MFs originating from circulating Ly6chigh mononuclear cells, which retain the CCR2 surface marker following differentiation [3].
The embryonic heart in the mouse reportedly contains two embryonic MF subgroups that can be subdivided based on CCR2, CCR2–MFs and CCR2+ MFs, derived from different lineages. During embryo growth, CCR2– and CCR2+ MFs occupied distinct positions of the embryonic heart with CCR2+ MFs located almost uniquely in the trabecular projections of the endocardium, while CCR2– MFs resided predominantly within the myocardial wall. The authors revealed the similar levels of expression of MF biomarkers CD64, MertK, and CD11c in CCR2– and CCR2+ MF subgroups were similar. CCR2+ MFs expressed the mononuclear cell marker Ly6c, while CCR2– MFs had more abundant expression of CX3CR1-GFP than CCR2+ MFs. Microarray gene expression profiling studies identified that CCR2– and CCR2+ MF subpopulations were different groups with distinctly different expression of 674 genes [36].
The adult human myocardium also contains a heterogeneous population of macrophages that can be partitioned into different subgroups on the basis of the expression of CCR2 and HLA-DR (human homolog of MHC-II): CCR2+HLA-DRhigh, CCR2–HLA-DRhigh MFs and CCR2+HLA-DRlow mononuclear cell. The authors subdivided MFs residing in the myocardium into CCR2+and CCR2– subpopulations with distinct tissue localization, origins, and properties. CCR2– and CCR2+ MFs displayed distinct gene expression profiles. CCR2+ MFs preferentially located within areas of heart scar tissue or fibrotic tissue in myocardium where they were inserted into areas containing type I collagen, while CCR2– MFs were closely linked to coronary endothelial cells [4].
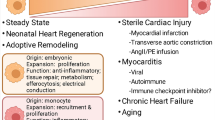
However, another study using flow cytometry with several cell surface biomarkers simultaneously with auto-fluorescence of cardiac MFs revealed the heterogeneity of cardiac-resident MFs. Most of the myocardial CD45+ cells were characterized as F4/80+CD11b+. In this heterogeneous subset, a similar examination identified four MF subgroups (R1–R4) in adult mouse heart. R1 was comprised of Auto+ Ly6c–, MHC-IIhigh, CX3CR1high, and CD206int. R2 was comprised of Auto+Ly6c–, MHC-IIlow, CX3CR1int, CD206high, and CD11clow. A small subset of R1 MFs were CD11chigh, while the majority of R1 were CD11clow, showing additional heterogeneity. R3 was comprised of Auto+Ly6c+, MerTK+, and CD206+-reserved Ly6c protein and accounted for approximately 2% of all the MFs in the heart. R4 was comprised of Auto–Ly6c+, MerTK–, and CD206– that represented monocytes. The R4 group existed in the Auto– gate, and could be distinguished from Ly6c+ MFs (R3) through the absence of MerTK alongside CD206. Among these four subsets of cardiac MFs, the primary cardiac MF populations were the R1 or R2 groups. These two MFs subsets featured more particles and more F4/80-expressing cells than mononuclear cells in the myocardium. Another study involving Ccr2GFP/+ mice (blood Ly6chigh monocyte sufficient) characterized all R4 cells (cardiac monocytes) and many CD11chigh MHC-IIhigh MFs (R1- CD11chigh) as CCR2+, while CD11clow (R1 and R2), and R3 (Ly6c+) MFs belonged to the CCR2–subset. R1-1 MFs were MHC-IIhighAuto+Ly6c–CX3CR1highCD206intCD11ChighCCR2+, R1-2 MFs were MHC-IIhighAuto+Ly6c–CX3CR1highCD206intCD11ClowCCR2–, R2 MFs were MHC-IIlowCX3CR1intCD206highCD11ClowCCR2–, and R3 MFs were Ly6c+MerTk+CD206+CCR2– and R4 mononuclear cells were Auto–Ly6c+MerTk–CD206–CCR2+ (Fig. 1). The primary cardiac MF populations were R1 or R2 groups [15].

Surface markers of cardiac macrophages defined in different developmental stages of mice and humans. *Surface markers in adult mice were derived from two articles
A recent study described that the normal adult murine heart includes four subsets of MFs, incorporating a group of cluster 1 (TIMD4+LYVE1+ MHC-IIlowCCR2–), cluster 2 (TIMD4–LYVE1– MHC-IIhighCCR2–) and two CCR2+MHC-IIhigh subsets, with gene expression differing in the subsets. The authors also found that CD45+CD64+CD14+ MFs could be sorted in the heart of adult patients suffering from cardiomyopathies. On the basis of HLA-DR (MHC-II) as well as the CCR2 biomarker, three subpopulations were identified: CCR2– HLA-DRhigh, CCR2+ HLA-DRhigh, and CCR2+ HLA-DRlow. These subsets appeared similar to mouse cardiac macrophages (Fig. 1) [14]. Another study revealed that a subgroup of resident Gata6 + pericardial macrophages are different from cardiac-resident macrophages. But this population exhibited transcriptional profiles similar to peritoneal and pleural cavity Gata6 + macrophages [11].
Ontogeny of cardiac- resident MFs
Findings made over the past decade have overturned the former perception of the genesis of cardiac MFs, by demonstrating that MFs residing in the heart are renewed by circulation blood mononuclear cells [26]. This view is appropriate for dermal and gut macrophages [39]. Parabiosis studies, in vivo cell tracking studies, data from bone marrow transplants, and genetic fate mapping have revealed the mixed ontological origins of several discrete cardiac-resident macrophage populations [39, 70].
MFs residing in the heart include a heterogeneous subset of populations that originate from different developmental lineages including adult mononuclear cells and embryonic cells. MFs originating from adult mononuclear cells are derived from definitive hematopoietic progenitor cells from the bone marrow and spleen. The cells subsequently infiltrate into the heart via certain signal pathways [16]. MFs originating from embryonic cells reside in the heart without mononuclear cell supplementation and are derived from distinct potential lineages containing primitive yolk sac, fetal liver monocyte progenitors, and recombination activating gene 1 (Rag1) + lympho-myeloid cells [36]. It was recently recognized that the embryonic heart includes several lineages of embryonic MFs with different roles that are vital for appropriate tissue growth. The majority of adult cardiac MFs originate from embryo precursors that migrated to the heart prior to birth and maintained themselves by in situ renewal, which was replenished in the absence of adult blood mononuclear cell pool [40, 70].
Yolk sac MFs and fetal liver mononuclear cells are embryonic macrophage precursors. Both can differentiate into cardiac-resident MFs and can arise from various progenitors external to, or within, an embryo. At embryonic day (E) 7.0, the posterior plate mesoderm in the blood islands of the extra-embryonic yolk sac (the inner or mesodermal layer of the yolk sac) gives rise to the first primitive hematopoiesis. Progenitors are generated at E7.25, which in turn give rise to primitive erythroblasts, megakaryocytes, alongside MFs [46]. The hemogenic endothelium of the yolk sac generates two erythro-myeloid progenitors (EMPs). The first, Blast Colony-Forming Cells, produce early Csf-1R+Myb-independent AA4.1–c-kit+ CD115+ EMPs (labeled at E7.5), which produce yolk sac MFs locally. The second, yolk sac Runx1+hemogenic endothelium, generates late multipotent Myb-dependent c-kit+AA4.1+EMPs (labeled at E8.5) that mostly migrate to the embryonic liver and then give rise to numerous lineages, containing fetal liver mononuclear cells that eventually produce tissue-retained MFs. The lineage potential and ability to produce mononuclear cell intermediates in the yolk sac and fetal liver of these temporally different EMPs is different [23]. Primitive EMPs differentiate in situ and primarily produce yolk sac MFs as well as red blood cells prior to their blood circulation. Early EMPs (primitive wave) in situ give rise mainly to yolk sac MFs in the absence of a mononuclear intermediate state. Late EMPs not only locally differentiate into yolk sac MF, but also transfer to the fetal liver once blood circulation is established. Furthermore, late EMPs (transient definitive wave) give rise to multiple lineages including fetal liver mononuclear cells, which in turn give rise to heart MFs [1, 28, 59].
We investigated the destiny of late EMPs from the yolk sac to the fetal liver. Over time, the late EMPs lost the ability of the early EMPs to generate yolk sac MFs. However, gradually they generated multiple myeloid progenitors in the fetal liver, which in turn produced granulocytes alongside mononuclear cells that ultimately generated tissue MFs. Another study revealed the molecular heterogeneity of two subsets was due to the differential expression of the colony-stimulating factor 1 receptor (CSF-1R) of early EMPs versus late EMPs. Early EMPs expressed CSF-1R messenger RNA, while late EMPs did not. On the other hand, the difference in the fate of EMPs’ fate in the lineage potential between the yolk sac and fetal liver was controlled by c-Myb. c-Myb is a transcription factor required for the proliferation and variation of each hematopoietic cell lineage. Primitive hematopoiesis develops independent of c-Myb because the earliest yolk sac progenitors (early EMPs) do not express c-Myb. However, late multipotent EMPs both express and rely on c-Myb. The growth of HSCs and CD11bhigh mononuclear cells and MFs requires the transcription factor Myb, while Myb is not essential for growth of yolk sac MFs as well as yolk sac-derived F4/80bright MFs in many organs, and cell subsets can persist in murine adults in the absence of hematopoietic stem cells [60].
In contrast, others described that during a short window of development in the early mouse yolk sac, the extra-embryonic mesoderm can contain three consecutive types of MFs, two of which were generated in situ. The authors described that first and instantaneous wave contained mature, maternal-derived MFs (CD45+ Mac-1+F4/80+CX3CR1–c-kit–) located in the mesoderm layer from 7.5 days postcoital (dpc) on. Maternally derived MFs participated in the elimination of pre-teratogenic cells, including radiation- or chemical-induced malformation. However, maternally derived MFs did not expand in culture. This effect was short-lived and these MFs were undetectable within yolk sac at 9.5 dpc. The second MF population is derived from yolk sac-monopotent MF-restricted precursors appearing in the yolk sac transiently prior to the erythro-myeloid precursors. These macrophage-restricted precursors (CD45– c-kit+) mature via a differentiation pathway (C: CD45–c-kit+, A1:CD45+ c-kit+ Mac1+, A2:CD45+c-kitlowCX3CR1low, A3:CD45+ c-kit–CX3CR1+F4/80+). These two waves were shown to be produced locally within the former yolk sac, independent of lympho-myeloid multipotent precursors (LMPs). So, the former “primitive MFs” represent a combination of the first instantaneous waves alongside definite yolk sac-monopotent precursors. Finally, the third wave of CX3CR1+ MFs are derived from the yolk sac-erythro-myeloid precursors (CD45– c-kit+). Both yolk sac-monopotent MF-restricted precursors and yolk sac-erythro-myeloid precursors displayed potent proliferative ability and can differentiate into Mac-1+ F4/80+ CX3CR1+ MFs. However, yolk sac-erythro-myeloid precursors differ from the yolk sac-monopotent MF-restricted precursors since the yolk sac-EMP precursors have fetal myeloid potential at the clonal level. Analysis of the gene expression profiles within myelomonocyte differentiation (MPO) revealed the expression of MPO from cell fractions A1 and A2 of yolk sac-monopotent precursors. The authors concluded that MFs derived from c-Kit+ CD45– precursors (both yolk sac-MP and yolk sac-EMP) progressed via an MPO+mononuclear cell period during their differentiation. Thus, they were associated with the “definitive” MF lineage [5].
Embryonic MF populations were established during three main successive phases. In mice, at embryonic age 7.0 (corresponding to 3weeks of gestation in humans), the first hematopoietic progenitors (yolk sac-derived monopotent progenitors) appeared in the yolk sac outside the embryo. These progenitors initiated “primitive hematopoiesis”, generating mostly yolk sac MFs as well as nucleated red blood cells [1, 28, 42, 59]. In both mouse and rat, yolk sac MFs first appeared at E9.0 in the blood islands of the yolk sac. Yolk sac MFs that developed instantly lacked any peroxidase activity and did not progress via the traditional mononuclear intermediate phase that is generally present in bone marrow [51, 63, 64]. From E8.5 to E10, yolk sac MFs (CD45+CD11blowF4/80highLy6C– CCR2–) were present in the yolk sac and were distributed within the embryo via the blood circulation. As a result, yolk sac MFs were evident throughout the body of the embryo proper at E10.5 and migrated to various tissues [41]. Primitive yolk sac MFs continued to locate in every organization from E10 to E12.5 without decreasing the proliferation ability [29]. Early in the development of the embryo, yolk sac MFs metastasized from the yolk sac to the embryonic heart, giving rise to a portion of the cardiac MFs. Yolk sac MFs were detected in the heart at E12.5 [28]. Another study reported that primitive yolk sac-derived MFs migrated to early embryo myocardium by approximately E9.5. The cells were readily evident by approximately E10.5. Yolk sac-derived MFs occurred within myocardium before the formation of fetal liver hematopoiesis. Yolk sac MFs of the heart comprised MHC-IIlow, CX3CR1high, F4/80high, CD11blow. These surface makers were similar to embryo yolk sac MFs in several other organs [15]. Low expression of CCR2 was evident on the surface of cardiac yolk sac MFs, described as the CCR2– subset. The embryonic yolk sac MFs initially seeded within the fetal myocardium through the mesothelium, a tissue within and just below the epicardium. The development of fetal yolk sac MFs in the myocardium requires the epicardium [61]. Intriguingly, yolk sac-derived MFs present in the adult myocardium is consistent with other MF subpopulations and a low, but vital, dedication of yolk sac MFs to adult MFs in the heart has been described.
Fetal mononuclear cells contribute to another population of adult MFs in the heart. Two waves of embryonic mononuclear cells have been demonstrated. The main wave comprises hematopoietic stem cell-independent progenitors derived mainly from yolk sac-originated late multi-lineage EMPs or LMPs. This generates many embryonic mononuclear cells between E12.5 and E17.5, which form another, definitive hematopoietic stem cell-dependent wave. The latter wave accounts for a small part of the subset of embryonic mononuclear cells after E17.5, the former being the major pathway arising from c-Myb+EMPs [21, 28].
From E8.0 to E8.25, EMPs and LMPs that emerge within hemogenic endothelium of extra-embryonic yolk sac are referred to as “second waves” or “transient definitive hematopoietic period”. Two types of EMPs appear within the yolk sac. The first “early’’ EMP-like cells (Csf-1R+Myb-independent AA4.1– c-kit+CD115+) at E7.5 primarily differentiate into MFs as well as on behalf of primitive progenitors. At E8.25, a second type of “late’’ EMPs (Myb-dependent c-kit+ AA4.1+) can differentiate into yolk sac MFs in situ, before finally migrating to fetal liver at E9.5 to produce progenitors with wider myeloid cell, including fetal liver mononuclear cells. These fetal liver mononuclear cells are also included in producing cardiac-resident MFs in adults [23]. With time, late EMPs lose their capacity to generate yolk sac MFs locally, while likely retaining the ability to differentiate into other lineages in the fetal liver that contain mononuclear cells. EMPs have also been observed in other hemogenic organs, for instance, the placenta and umbilical cord. From E9.0 or E9.5, they migrate to seed and colonize the fetal liver after circulating in the blood. It is not clear whether early and late EMPs are different progenitor cell groups derived from distinct sources, or the same subset that exists along a continuum of maturation periods [21].
Eventually, the intra-embryonic mesoderm commits to the hematopoietic lineage and novel types of hematopoietic progenitors concurrently emerged, with the almost simultaneous appearance of late EMPs at E8.5. The para-aortic splanchnopleure (P-Sp) can generate pre-hematopoietic stem cells (immature hematopoietic stem cells). At E10.5, the hematopoietic endothelium of the aorta–gonado-mesonephros region gives rise to embryonic mature hematopoietic stem cells. Pre-hematopoietic stem cells as well as mature hematopoietic stem cells migrate to and colonize the embryonic liver, where they expand and differentiate starting from E12.5, to finally establish “definitive hematopoiesis” [71]. The fetal liver becomes the primary hematopoietic tissue from E11.5. It produces all hematopoietic lineages, including the monocyte-derived macrophages, and enlarging the definitive hematopoietic stem cell group prior to their infiltration into the spleen alongside the bone marrow. Thus, in the embryonic liver, a series of yolk sac late EMP-derived hematopoiesis, followed by hematopoietic stem cell-originated hematopoiesis. These precursors then colonize the embryonic bone marrow where these cells ultimately produce adult bone marrow hematopoietic stem cells. Hematopoietic stem cells derived from bone marrow dominate hematopoiesis and generate all immune lineages only during the perinatal stage [23, 29].
Usually mononuclear cells generated in adult bone marrow-derived successively from MF-dendritic cell precursors (MDPs) as well as common monocyte progenitors (cMoPs). A study that investigated the origin of fetal liver mononuclear cells detected a differentiation continuum of progenitors in the fetal liver. The continuum included Lin– cKit+ Flt3+ CSF– 1R+Ly6c– MDP progenitors within fetal liver present from E12.5 to E16.5, as well as Lin– cKit+ Flt3–CSF-1R+ Ly6c+ myeloid progenitors (MPs). The phenotype in Ly6c+ MP group was similar to that in cMoP. By comparison, the Ly6c– group in myeloid progenitors disappeared in adult bone marrow, indicating that it might be an instantaneous fetal MP subset specifically within the fetal liver. The authors designated these subsets as fetal liver cMoP, fetal liver MP, and fetal liver MDP. Fetal liver MPs and fetal liver MDPs were mostly closely related. FL cMoP (Lin–cKit+Flt3–CSF-1R+Ly6c+) seemed to be an intermediate among the two subgroups and embryonic mononuclear cells. The c-Myb+ EMPs gave rise to the fetal liver MP, while the LMPs gave rise to the fetal liver MDPs, which in turn generated fetal liver cMoP (CCR2low CX3CR1low). Finally, the latter gave rise to the fetal liver mononuclear cells and MFs. The authors suggested the occurrence of donation from Flt3-dependent MDP to embryonic mononuclear cells alongside MFs. Embryonic mononuclear cells arose through an FLT3-, and perhaps also an HSC-independent pathway (fetal liver MPs). Instantaneous fetal liver MPs, probably the progeny of c-Myb+ EMPs, are the likely primary origin of embryonic mononuclear cells [28].
Fetal mononuclear cells have been shown to emerge within the fetal liver at approximately E12.5 as two distinct Ly6c expression groups, similar to mononuclear cells in adult bone marrow. Both embryonic mononuclear cell groups express CCR2 (CCR2+), even though the expression is not necessary for their transfer from the fetal liver or infiltration into organs. Compared with mononuclear cells in adult bone marrow, expression of the chemokine receptor CX3CR1 begins in embryonic mononuclear cells only in the blood, after their transfer from the fetal liver. Embryonic mononuclear cells display the increased expression of CX3CR1 and the macrophage biomarkers CD64 and Mertk, and decreased expression of Ly6c after tissue penetration, as the cells differentiate into tissue MFs. At approximately E12.5, embryonic mononuclear cells in the fetal liver give rise to macrophages in situ and then move to the heart around approximately E14.5. MFs originating from fetal mononuclear cells include CD45+, CX3CR1low, F4/80low, CD11bhigh, CCR2+, and Ly6c+. The early tissue MFs declined in number from E10.5 to E16.5, By E16.5, embryonic mononuclear cells are the primary myeloid cell subset in the organs.
Embryonic mouse heart contains CCR2– and CCR2+ MFs, and CCR2– MFs overexpress transcripts related to yolk sac-derived and -resident macrophage subgroups (CX3CR1, Lyve1, Emr1, Cd207, Ccl12). However, expression of mononuclear cell-originating MF transcripts was elevated in the CCR2+ subpopulation. These transcripts included Ly6c, CxCR2, Sell, Lrf5 and Nr4a1. The authors similarly divided the embryonic cardiac MFs into F4/80highCD11blow and F4/80lowCD11bhigh subsets and reported that the F4/80highCD11blowsubset exclusively consisted of CCR2–Ly6c– MFs, and so were derived from yolk sac progenitors. The F4/80lowCD11bhigh subset predominately contained CCR2+Ly6c+ MFs, consistent with fetal mononuclear cell origin. The authors further used genetic lineage tracing and immunostaining analysis to demonstrate that CCR2–MHC-IIlow MFs originated from yolk sac progenitors, while CCR2+MHC-IIlowMFs mainly originated from definitive hematopoiesis (predominantly embryonic mononuclear cell progenitors), with only a minority of this subset originating from Rag1+lympho-myeloid progenitors. CCR2–MHC-IIlow and CCR2+MHC-IIlow MFs migrated into the myocardium at distinct times during development. CCR2–MHC-IIlow MFs were first detected in cardiac tissue at E12.5. Subsequently, the cells populated the subepicardial space and persisted within the ventricular myocardium throughout development. From E13.5 to E14.5, yolk sac-derived CCR2– MFs played a vital role in growth during the maturation of the coronary system. In contrast, at E14.5, CCR2+MHC-IIlow MFs were observed within the endocardium trabeculae. The function of these CCR2–MHC-IIlow MFs is vague because they are dispensable for the development of the heart [36].
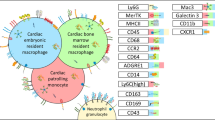
When adult hematopoietic stem cells give rise to all lineages containing myeloid, lymphoid, megakaryocyte, and erythroid cells, they transiently pass through the FLT3+stage. However, FLT3 may not be used to differentiate primitive variety from definitive hematopoietic MF originations in the developing stage. The authors suggested that most adult MFs in the heart develop through FLT3-independent pathways and contain an ontological subset that is comprised of MFs that originated from the fetal yolk sac and from fetal liver mononuclear cells. The suggestion reflects the finding that the tissue-resident MFs were established during embryonic development at a time when definitive hematopoiesis could not efficiently drive FLT3-mediated recombination in the fetal liver. The authors further revealed that the F4/80highCD11blow yolk sac-derived MFs consisted of FLT3-Cre–, while MFs that originated from FL mononuclear cells (F4/80low CD11bhigh) comprised only 5% of FLT3-Cre+ (5%) MFs in all organs tested. These findings revealed that penetration of hematopoietic stem cell-derived mononuclear cells had started. The collective findings indicate that at primitive MFs constitute embryonic yolk sac-derived MFs (FLT3-Cre negative) and definitive MFs are subdivided into embryonic fetal liver monocyte-derived MFs (FLT3-Cre negative) and adult-derived MF subpopulations (FLT3-Cre positive). Figure 2 depicts the different origins of the cardiac-resident macrophages during embryonic development stages.

There are different originations of the cardiac-resident macrophages (MFs) during the stages of embryo development. MF embryonic precursors include yolk sac macrophages (YS MFs) and fetal liver monocytes (FL MOs) arose from various progenitors at different regions exterior to, or inside, the embryo. They contributed to cardiac-resident MFs. From embryonic day (E) 7.0 to 9.0, MFs are seeded from yolk sac precursors; during E12.5 to 17.5, monocyte-like cells are capable of seeding macrophages from the fetal liver. From E17.5 through adulthood, mature monocytes from bone marrow are able to seed a variety of MF lineages in the tissues
Renewal of cardiac-resident MFs
MFs residing in the heart can participate in immunity, tissue repair, and tissue homeostasis of the heart. The long-held notion was that MFs residing in the heart renew themselves from recruitment of circulating blood mononuclear cells during steady state and following cardiac damage. However, accumulating evidence has established that the composition of the cardiac MF pool is not static [20]. Rather, the composition of the cardiac-resident MF population changes during postnatal development and the renewal of cardiac-resident MFs depend on the state of the heart along with age.
Most hematopoietic cells replenish from adult hematopoietic stem cells. However, the commitment of adult hematopoietic stem cells to MFs residing in some tissues varies among origins and is augmented with age. MFs within some adult organs, including the brain, epidermis, lung, and liver, can self-replenish in the absence of hematopoietic stem cells [70]. Several other organs display rapid steady-state infiltration like gut and dermis. However, MFs in the heart and pancreas are open organs with moderate steady-state infiltration [23].
During the steady state, MFs resident in the heart can expand locally in the heart and renew via in situ proliferation, with relatively little contribution of blood monocytes to cardiac MFs in the healthy myocardium. During the steady state, there is some substitution of the resident CD11clow MHC-IIhigh and MHC-IIlow MFs by blood mononuclear cells. CD11clow macrophage subsets (R1 and R2) mainly exist in the absence of imported circulating mononuclear cells and can replenish through local expansion. CD11chigh MHC-IIhigh macrophages (R1- CD11chigh) are only modestly substituted by circulating mononuclear cells, showing that these subsets are either derived in situ or macrobiotic. Substitution partially involves Ly6c+ MFs (R3), but not total substitution by mononuclear cells. CD11chighMHC-IIhigh (R1-CD11chigh) MFs and Ly6c+ MFs renew through both in situ proliferation and by mononuclear cell substitution to various degrees [15]. Another study also revealed that during steady state, most CCR2+ MFs (> 80%) in the myocardium and a lower fraction of MHC-IIhigh MFs (approximately 25%) residing within cardiac tissue are substituted by circulating monocytes, while only approximately 12% of the replacement involves MHC-IIlow MFs [14].
Interestingly, in mice, the cardiac-resident MFs experience dynamic shifts over time, with an age-related stepwise substitution of fetal-origin heart-resident MFs by mononuclear cell-origin MFs occurring during normal postnatal development, even in the absence of inflammation or injury. The authors described that all embryo-derived heart MFs were MHC-II– at birth, but that, after several weeks, four subpopulations had emerged with a progressive increase of MHC-II+ heart MFs and a decrease of CX3CR1+ heart MFs. The authors further described that a remarkable fraction of heart MFs established during early embryo growth persists at birth, with an eight- to tenfold decrease of the most proliferative embryo-derived MFs (CX3CR1+MHC-II– heart MFs) and total heart MFs with age. A remarkable decline in the proliferation rate and self-renewal capacity of the heart-resident embryo-derived MFs was evident with age, which led to the gradual reduction in the self-renewal of the pool of cardiac-resident MFs and fewer proliferating MFs within adult hearts. Whether the decline of this capacity is permanent or can be reactivated is unclear. The authors also described that the age-related decrease of embryo-derived MFs was substituted by monocyte-derived MFs. Infiltrating Ly6c+monocytes could differentiate into all subsets of heart MFs, which preferentially contributed to MHC-II+ MFs and led to a varied distribution of the resident MF pool over time. Additionally, the serial augmentation of MHC-II+ and CX3CR1–MFs along with the reduction in the number of CX3CR1+ MHC-II– cells were evident [44]. Another study also found an age-associated variation in composition, gene profile, and function of MFs residing in the heart. The proliferative capacity of the MFs decreased with age and the expression of the Cx3cr1 gene was lost in cardiac-resident MFs. These events induced age-associated variation in the distribution of MFs and gene expression. Age-dependent cardiac-resident MFs show a reduced expression of immunoreactive genes (Cx3cr1, Lpar6, CD9, Cxcr4, Itga6, and Tgfβr1), and an increased expression of fibrogenic genes (Ltc4s, Retnla, Fgfr1, Mmp9 and Ccl24) prior to cardiac functional impairment [53].
However, in the human heart, CCR2–HLA-DRhigh MFs comprise a tissue-resident subset that is exclusively replenished by cell proliferation in situ. In contrast, CCR2+HLA-DRhighMFs are maintained by a combination of infiltrating mononuclear cells as well as cell proliferation. The turnover rates in CCR2+HLA-DRhigh MFs are higher than the CCR2–HLA-DRhigh MFs [4].
The homeostasis of the heart can be perturbed following the depletion of MFs, angiotensin II (Ang-II) infusion, severe inflammation, sub-lethal irradiation, and myocardial infarction. In one study, when cardiac monocytes and MFs are depleted with the injection of clodronate liposomes, circulating Ly6chigh monocytes could readily replenish the cardiac MF pool and differentiate into all heart-resident MF subsets. In contrast, CD11chighMHC-Ihigh MFs and Ly6c+ MFs appeared to renew both by in situ proliferation and mononuclear cell substitution to varying degrees. These results correlated well with observations made after the induction of myeloid cell depletion in CD11bDTR/+ mice. The authors also explored the inflammatory effects of Ang-II on cardiac MF populations. Ang-II can induce the rapid recruitment of Ly6c+ monocytes. These monocytes could differentiate into all MF groups. In addition to the influx of monocytes, MFs were maintained through local proliferation when the heart suffered inflammation. However, the cardiac-resident MFs (CCR2–CD11clowLy6c–) proliferated independent of mononuclear cell supplementation, but through in situ expansion, while mononuclear cells expressing CCR2 could differentiate into both CCR2+ and CCR2– MF subpopulations by infiltration and subsequent expansion [15]. After myocardial infarction, an abrupt and almost absolute extinction of cardiac MFs was observed within the ensuring 24 h during myocardial ischemia in the injured myocardium. The number of dead MFs significantly and rapidly increased within the first 2 h after injury. Then bone marrow-derived mononuclear cells overwhelmingly entered the pool of heart MFs residing in acute infarctions and proliferated. Ly-6Chigh mononuclear cells infiltrate the injured heart due to the marked increase of monocyte-chemoattractant protein-1 (MCP-1) within the early inflammation stage of infarct healing [13]. A small number of Ly-6Clowmononuclear cells can infiltrate through CX3CR1 during the second stage [27]. Many Ly-6Clow mononuclear cells can be recruited within the 2 weeks following cardiac ischemia. After this time, MFs residing in infarct can be restored independent of the mononuclear cell blood pool and instead again depend on local expansion, similar to the proliferation that occurs during the steady condition [27].
Functions of cardiac-resident MFs
Studies have mainly targeted the functions of MFs that originate from circulating monocytes on the reconstruction of the heart. MFs comprise the major part of the innate immune system, which is important in many pathophysiological mechanisms and cardiac diseases [7, 19, 34, 49], such as atherosclerosis [45, 57], myocardial infarction [14, 27, 47], heart failure [18], and myocarditis [37]. Recently, one study revealed that pericardial Gata6 + macrophages contribute to cardiac repair upon injury and prevent cardiac fibrosis, highlighting the complexity of the cardiac macrophages [11].
Our understanding about features of cardiac-resident MFs remains limited. As regards their importance in immune functions, MFs participate in phagocytosis along with antigen presentation. MFs defend against host cell damage and pathogens like bacteria [37]. Knowledge of the functions of cardiac MFs is still developing. MFs residing in the heart and infiltrating MFs originate from different sources during development. They also display different patterns of gene expressions and functions. Transcriptional profiling of CCR2+and CCR2– MFs has revealed that genes within CCR2+ macrophages are related to inflammation signaling involving tumor necrosis factor and nuclear factor-kappa B, inflammation, allograft rejection, and several inflammatory signaling pathways, including interleukin 2/signal transducer and activator of transcription 5 (IL2/STAT5), IL6/STAT3, interferon-gamma, hypoxia and K-RAS. By comparison, CCR2– MFs express genes related to epithelial–mesenchymal transition, coagulation, myogenesis, p53, and IL2/STAT5 signals [4]. Gene expression and pathway analyses of cardiac-resident CCR2–MFs have revealed that the cluster TIMD4+LYVE1+MHC-IIlowCCR2– pathway is related to homeostasis sas well as regenerative capabilities, for example, endocytosis, lysosome-mediated degradation, and angiogenesis. Higher expression of antigen presentation has been observed in the MHC-II cluster (TIMD4+LYVE1–MHC-IIhighCCR2–). Translational-ribosomal pathways also significantly accumulate in the MHC-II cluster (TIMD4+LYVE1–MHC-IIhighCCR2–), CCR2 cluster (CCR2+MHC-IIhigh) and Isg cluster (CCR2+MHC-IIhiIrf7–,Isg20+,Ifit1+), while classic inflammatory pathways are enriched in the CCR2 cluster (CCR2+MHC-IIhigh) and Isg cluster (CCR2+MHC-IIhighIrf7+, Isg20+, Ifit1+) [14].
This section discusses the different functions of cardiac-resident CCR2–MFs and CCR2+MFs.
Function of cardiac-resident CCR2–MFs
Cardiac-resident CCR2–MFs play a vital role in advancement and maturation of coronary tissue
Embryonic cardiac development includes a complicated set of fetal MF groups, which can be subdivided into CCR2– and CCR2+MFs. The distinct lineages of embryonic MF subsets have differing functions. CCR2–MFs derived from the yolk sac are pro-angiogenic so they play a vital role in coronary vascular growth and maturations. By facilitating the alternative expansion of perfused vasculature, these MFs participate in reconstruction of the primitive coronary plexus. CCR2–MFs do not participate in this reconstruction in the embryonic heart. Instead, these cells are critical in the reconstruction of the vascular plexus into a mature vascular tree that includes vessels of different widths. In particular, MFs preferentially facilitate the development of perfused blood vessels. The trade-off is that the blood vessels receive little or no perfusion. Mechanistically, CCR2–MFs directly or indirectly regulate coronary endothelial cell proliferation and regulate coronary remodeling in a potentially flow-dependent manner. IGF associated with CCR2–MF may alternatively stimulate expansion of the perfused coronary vasculature by promoting the infiltration of endotheliocytes into locations of vascular perfusion and consecutive union of endotheliocytes into regions of active reconstitution. By contrast, CCR2+MFs seem to be dispensable for heart development [36].
Role of cardiac CCR2–MFs in cardiac electrophysiology
MFs residing in the heart have a spindle-like appearance and a speckle appearance among cardiac muscle cells, smooth muscle cells, and endotheliocytes in the myocardium. The sino-atrial node in the heart can engender an electrical impulse that is continuously transmitted to activate the atria, atrio-ventricular node (AVN), His and Purkinje systems, and finally the ventricles. The AVN is located at the base of the right atrium. This site contains cardiomyocytes with distinct action potentials that maintain the unique electrical conduction between the atrial and ventricle chambers, which is essential in the cardiac conduction systems. One study that examined MFs located in the mouse and human distal AVN regions showed that the mouse AV node contains more macrophages than the LV-free wall. Many CD45+white blood cells within the murine myocardium are CD11b+F4/80+Ly6ClowMFs. This indicates that circulating cells contributed marginally to MFs within the AV node in parabiosis study, consistent with MFs in the LV-free wall. These MFs contribute to cardiac conduction. RNA-seq and real-time PCR analyses demonstrated AV node and LV-free wall MF expression channels as well as ion exchangers. Microarray data revealed an abundance of genes related to electrical conduction in heart MFs. The gene expression profile in the mouse AV node MFs resembled the genes within MFs residing in the ventricular region. These MFs express genes that were associated with cardiac electrophysiology. These AVN MFs mainly expressed Cx43 and Cx43-containing gap junctions that connect the MFs and cardiomyocytes. This leads to cyclical MF depolarization and action potentials in MF that are synchronized with action potentials of directly connected cardiomyocytes. AVN MFs can modulate electrophysiological properties of cardiomyocytes, decrease the effective refractory period of the AVN conduction structure and facilitate AVN conduction. The electrical load of MFs depolarizes resting cardiomyocytes, which contributes to a reduced action potential upstroke velocity and overshoot, and faster early repolarization. Deleting Cx43 in MFs or elimination of MFs using a CD11b-DTR model delayed AV conduction and led to an AV block. The authors suggested that MFs within the AV node engage in further, Cx43-independent tasks, which might or might not be associated with conduction. These surprising results extend our understanding of the donations of MFs to physiological cardiac conduction as well as anomaly of heart rhythm [30].
Based on the above observations, MFs may be involved in the occurrence and development of atrial fibrillation (AF). A relationship between inflammatory and AF has been described [6, 17, 25, 52]. Numerous inflammation biomarkers and mediators, for instance, tumor necrosis factor-alpha, C-reactive protein, several IL family molecules (IL-2, IL-6, IL-8) as well as monocyte chemoattractant protein (MCP)-1 are significantly increased in the atria following atrial fibrillation. These biomarkers have been related to the initiation or result of atrial fibrillation [24, 52]. Whether inflammation is an outcome of, or a reason for, atrial fibrillation is unclear. The effect of inflammatory in the beginning of atrial fibrillation was determined mainly based on the phenomenon that several inflammation conditions, for example, myocarditis, pericarditis, and rheumatoid arthritis, as well as cardiac surgeries, were related to atrial fibrillation [35]. Histological analyses conducted in a mouse model of atrial fibrillation and in patients with atrial fibrillation revealed an augmented amount of mononuclear cells and MFs in atrial tissue compared with control group. In some clinical studies, administration of glucocorticoids reduced the recurrence of atrial fibrillation and depletion of MFs, while clodronate liposomes can reduce the inducibility of atrial fibrillation in a murine model [12, 62]. However, the inflammatory pathway underlying atrial fibrillation activated by the immune system leukocyte or the cardiac-resident MFs remains unclear. Whether inflammation associated with atrial fibrillation is a systemic or regional phenomenon is also unclear [25]. More studies that focus on the cardiac-resident macrophages in the physiopathologic mechanism of atrial fibrillation are needed.
Cardiac-resident CCR2–MFs can promote heart regeneration in neonate mice
In neonatal mice, after cryo-ablation, myocardial infarction, and other heart injuries, cardiomyocytes can proliferate effectively, which contributes to the regeneration of the newborn mouse heart without scar formation. However, this regenerative ability disappears after postnatal day 7 [24]. Depletion of macrophages impaired the regeneration capacity of neonatal mice heart. The results indicate that embryo-derived MFs play crucial roles in neonate mice heart repair following heart injury, because the newborn murine proliferate a subset of embryo-derived cardiac-resident MFs, which produce a minor inflammatory reaction, and then facilitate the heart repair through the stimulation of coronary angiogenesis and expansion of myocardial cell. In response to injury, the newborn cardiac cells selectively expanded the amount of MHC-IIlowCCR2–MFs and do not recruit more CCR2+ monocytes. These MHC-IIlowCCR2– MFs represent a relatively pure embryo-derived MF subset. In contrast, following ischemic injury in the adult heart, the embryo-derived MFs are replaced by the recruited monocytes and MHC-IIhighCCR2+ monocyte-derived MFs which are pro-inflammatory and lack reparative activities, resulting in the mediation of scar formation rather than regeneration [2, 33].
A recent study sought to uncover the molecular mechanisms underlying neonatal heart regeneration. The researchers examined the transcriptomes and epigenomes in regenerative and non-regenerative mouse hearts over a 7-day period following myocardial infarction injury. The author identified genes and biological processes that were uniquely activated in injured regenerative hearts, a unique immune response in regenerative hearts, and retained embryonic cardiogenic gene program active during neonatal heart regeneration. The authors further demonstrated that injury of the regenerative heart triggered a unique immune response involving regeneration promoted by the macrophage-secreted cytokine factor, CCL24, and Igf2bp3, which encoded an RNA-binding protein and enhanced regeneration. Compared with P14 macrophages, cytokine factor Ccl24 is preferentially expressed in P1 macrophages. However, the mechanism of Ccl24 on cardiac regeneration and its downstream signal pathway still need to be studied. Therefore, reactivation of the developmental and cell cycle gene programs in adult cardiomyocytes serves as a potential therapeutic approach to replenish lost cardiomyocytes after ischemic injury [67].
Role of cardiac-resident CCR2– macrophages in the inhibition of recruitment of monocytes during heart injury
In a variety of murine models of cardiomyocyte injury, such as myocardial infarction, reperfusion myocardial infarction and diphtheria toxin cardiomyocyte ablation, a change in MF development has been demonstrated, in which infiltrating CCR2+Ly6chigh mononuclear cells and CCR2+mononuclear cell-originated MFs primarily substituted for MFs residing in the heart. A mass of monocytes and monocyte-derived MFs migrate to the myocardium and accelerate harmful reconstruction as well as the pathogenic mechanism of heart failure. Nevertheless, the recruitment and fate of circulating monocytes and MFs are differentially orchestrated by distinct subsets of cardiac-resident CCR2+ and CCR2– MFs after myocardial injury. CCR2+ MFs in the heart facilitate mononuclear cell migration depending on myeloid differentiation primary response 88(MYD88), which leads to the generation of MCPs and activation of mononuclear cells. CCR2+MFs residing in the heart are critical drivers of monocyte recruitment, fate specification, inflammation, and adverse LV remodeling. In contrast, cardiac-resident CCR2–MFs can inhibit monocyte recruitment and depletion of cardiac-resident CCR2–MFs before myocardial infarction results in significant shifts in the fact of monocyte specification, augmentation of MF proliferation, expanded infarct size, decreased LV contractive function, as well as exaggerated reconstruction of left ventricle. RNA-seq of distinct MF subsets has revealed that compared with cardiac-resident CCR2–MFs, cardiac-resident CCR2+MFs also represent an inflammatory population. Cardiac-resident CCR2–MFs differentially expressed several growth factors (Igf1 [insulin-like growth factor 1], Pdgfc [platelet-derived growth factor C], Hbegf [heparin-binding epidermal-like growth factor], and Cyr61 [cysteine-rich angiogenic inducer61]) and genes associated with myogenesis, DNA repair, epithelial–mesenchymal transitions, and RAS signaling [3].
A myocardial ischemia reperfusion study revealed that MerTK (MER proto-oncogene tyrosine kinase)-dependent phagocytic clearance of MHCIIloCCR2– cardiac-resident MFs ameliorated cardiac damage after myocardium injury. However, the capacity of MerTK in the heart was destroyed by MerTK cleavage caused by reperfusion. MerTK was significantly higher on MHC-IIlowCCR2– cardiac-resident MF and was required for both MHC-IIlowCCR2–MFs conservation and generation of anti-inflammatory mediator. MHC-IIlowCCR2–MFs played a critical role in the repairing of the heart through MerTK after ischemia reperfusion injury. The findings indicated that the trigger of MerTK cleavage is the CCR2-dependent infiltration of circulating Ly6chigh monocytes [10].
Cardiac-resident CCR2–MFs limit adverse LV remodeling and dysfunction
A study used a Cx3cr1CreER−YFP:R26Td/DTR murine model to mark resident MFs and efficiently exhaust absolute CCR2–MFs in the heart, in the absence of influence of other cell types. The authors found that diphtheria toxin injection alone during the steady state without any detectable infiltration of neutrophil granulocytes and MFs did not result in myocardial fibrosis in the wild type. However, exhaustion of MFs residing in the heart led to the deterioration of the global left ventricular contraction capacity between day 7 and day 28 post-infarction, with the increase of cardiac hypertrophy, fibrosis and increased late mortality by day 35 post-infarction in diphtheria toxin-treated Cx3cr1CreER−YFP:R26Td/DTR mice compared to Cx3cr1CreER−YFP:R26Td/ + mice. However, comparison of the magnitude of the inflammation response in MF-depleted mice with non-depleted (Cx3cr1CreER−YFP: R26Td/ +) mice at day 5 post-infarct revealed no remarkable variation. The findings indicated revealed that removal of resident MFs did not lead to an exaggerated inflammation reaction [14].
Function of cardiac-resident CCR2+MFs
Role of CCR2+MFs in the recruitment of leukocytes (monocyte and neutrophil) during heart injury
As described above, during the death of cardiomyocytes, CCR2+ MFs residing in the heart stimulate the infiltration of mononuclear cells into the myocardium. This depends on the MYD88 protein, which leads to the secretion of MCPs as well as mononuclear cell excitation [43]. These events are critical drivers of mononuclear cell recruitment, fate specification, inflammation, heart failure pathogenesis and adverse heart remodeling [3].
In addition to mononuclear cells, neutrophil granulocytes also have an important impact on myocardial ischemia/reperfusion that can occur rapidly in injured myocardium [9]. Once they have infiltrated, neutrophil granulocytes release a variety of inflammation mediators and chemotactic moderators that result in cell damage or assist in infiltrating of other immune cells [31]. The authors showed that during ischemia/reperfusion, cardiac-resident CCR2+monocytes and cardiac-resident CCR2+monocyte-derived macrophages facilitated the permeation of neutrophils into injured hearts by a Toll-like receptor 9/MyD88-dependent generation of the chemotactic mediators CXCL2 and CXCL5 [38].
Pro-inflammatory role of CCR2+ MFs
The analysis of gene expression, chemotactic mediators, inflammation moderators, cytokines as well as related signal pathways of the CCR2+ MFs revealed that these MFs represented an inflammatory macrophage population. CCR2+ MFs express many chemotactic mediators, chemokine receptors, and inflammatory factors associated with IL-1, IL-6, and nuclear factor-kappa-B signals. In vitro, compared to CCR2– MFs, following stimulation with lipopolysaccharide, CCR2+ MFs express higher levels of the pro-inflammatory factors: IL-1b and CCL7. Human or genotypic slice culture systems also revealed that human heart--resident CCR2+ MFs are pro-inflammatory [4].
Role of CCR2+MFs in LV dysfunction
After cardiac mechanical unloading, CCR2+ MFs residing in the heart have been correlated with LV contraction function that occurs with heart remodeling. Using immunostaining, the authors found that the numbers of CCR2+ MFs and their percentage were associated with LV contractility following left ventricular assist device (LVAD) implantation. Patients with improved left ventricular contractility 6 months following LVAD implantation had lower numbers and percentage of CCR2+MFs both during LVAD implantation and at the time of explant [4].
Cardiac CCR2+ and CX3CR1+ MFs can improve heart function in cardiac stem-cell therapy in mice after ischemia/reperfusion
Cell therapy with adult stem cells (primary bone marrow mononuclear cells, MNCs and c-Kit cardiac mesenchymal cells/cardiac progenitors cells, CPCs) to regenerate damaged heart tissue continue to be extensively used in human clinical trials and animal studies [56]. However, the mechanistic basis for this therapy remains unclear. Recently, one study revealed that the benefits of cardiac stem-cell therapy using MNCs and CPCs were not associated with new cardiomyocyte and cardiac endothelial cell formation. This stem-cell therapy in mice after heart ischemic injury could significantly improve cardiac function through an acute sterile immune response characterized by the short-lived and local accumulation of CCR2+ and CX3CR1+ macrophages. The protective effect of this stem-cell therapy disappeared when the acute immune response was suppressed or the macrophages were eliminated. CCR2 null mice and mice lacking CX3R1 could not be benefitted from cardiac stem-cell therapy. However, CCR2 deficiency in wild C57BI/6J mice significantly improved heart function after ischemia/reperfusion. In contrast, CX3CR1−/− mice showed cardiac dysfunction after ischemia/reperfusion injury. In mechanism, injection of MNCs into mice after ischemia/reperfusion changed cardiac fibroblast activity-reduced border zone extracellular matrix content [65]. And at the same time, another study found that pericardial Gata6 + macrophages benefit cardiac repair upon injury and prevent cardiac fibrosis, highlighting the complexity of the functions of cardiac macrophages in the heart injury [11].
Conclusions
In summary, our review suggests the heterogeneity of cardiac in situ macrophages and they play a vital and complicated role in both physiological and pathological processes of the heart, and are expected to become new targets for the treatment of cardiovascular diseases in the future. In terms of quantity, cardiac-resident macrophages account for the largest proportion of all leukocytes in the heart and their classifications and sources are diversified and complicated. In the hearts of adult mice and humans, multiple subpopulations of macrophage cells co-exist; in addition to adult bone marrow-mononuclear-derived macrophages, there also exist embryonic EMP-derived macrophages and embryonic mononuclear-derived macrophages, and their self-renewal and proliferation patterns also dynamically changed depending on ageing and different situations. In terms of function, with the deepening of various studies, we found that the cardiac-resident macrophages play a far more important role in cardiovascular disease than their role as immune cells. Different cardiac-resident macrophages play a different role in neonatal cardiac regeneration, cardiac remodeling and cardiac electrophysiology through diverse mechanisms. At present, with the development of single-cell sequencing and transcriptome sequencing based on different classification of cardiac-resident macrophage cells, we have found that there are more detailed classifications and functional differences between different types of cardiac-resident macrophages. In the future, we also require high-resolution biomarkers of different cardiac-resident macrophages to study them. Based on the results of single-cell sequence, we might carefully identify different cardiac-resident macrophages that play a role in various cardiovascular diseases, and carefully study their physiological characteristics as well as their functions, which are expected to provide new therapeutic strategies for cardiac diseases and precision medicine in the future.
References
Auerbach R, Huang H, Lu L (1996) Hematopoietic stem cells in the mouse embryonic yolk sac. Stem Cells 14:269–280. https://doi.org/10.1002/stem.140269
Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, Sadek HA, Olson EN (2014) Macrophages are required for neonatal heart regeneration. J Clin Invest 124:1382–1392. https://doi.org/10.1172/jci72181
Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, Mohan J, Ivey B, Hsiao HM, Weinheimer C, Kovacs A, Epelman S, Artyomov M, Kreisel D, Lavine KJ (2019) Tissue resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res 124:263–278. https://doi.org/10.1161/circresaha.118.314028
Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M (2018) The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med 24:1234–1245. https://doi.org/10.1038/s41591-018-0059-x
Bertrand JY, Jalil A, Klaine M, Jung S, Cumano A, Godin I (2005) Three pathways to mature macrophages in the early mouse yolk sac. Blood 106:3004–3011. https://doi.org/10.1182/blood-2005-02-0461
Boos CJ, Anderson RA, Lip GY (2006) Is atrial fibrillation an inflammatory disorder? Eur Heart J 27:136–149. https://doi.org/10.1093/eurheartj/ehi645
Coverstone ED, Bach RG, Chen L, Bierut LJ, Li AY, Lenzini PA, O'Neill HC, Spertus JA, Sucharov CC, Stitzel JA, Schilling JD, Cresci S (2018) A novel genetic marker of decreased inflammation and improved survival after acute myocardial infarction. Basic Res Cardiol 113:38. https://doi.org/10.1007/s00395-018-0697-7
Dagenais GR, Leong DP, Rangarajan S, Lanas F, Lopez-Jaramillo P, Gupta R, Diaz R, Avezum A, Oliveira GBF, Wielgosz A, Parambath SR, Mony P, Alhabib KF, Temizhan A, Ismail N, Chifamba J, Yeates K, Khatib R, Rahman O, Zatonska K, Kazmi K, Wei L, Zhu J, Rosengren A, Vijayakumar K, Kaur M, Mohan V, Yusufali A, Kelishadi R, Teo KK, Joseph P, Yusuf S (2019) Variations in common diseases, hospital admissions, and deaths in middle-aged adults in 21 countries from five continents (PURE): a prospective cohort study. Lancet. https://doi.org/10.1016/s0140-6736(19)32007-0
Daseke MJ 2nd, Valerio FM, Kalusche WJ, Ma Y, DeLeon-Pennell KY, Lindsey ML (2019) Neutrophil proteome shifts over the myocardial infarction time continuum. Basic Res Cardiol 114:37. https://doi.org/10.1007/s00395-019-0746-x
DeBerge M, Yeap XY, Dehn S, Zhang S, Grigoryeva L, Misener S, Procissi D, Zhou X, Lee DC, Muller WA, Luo X, Rothlin C, Tabas I, Thorp EB (2017) MerTK Cleavage on resident cardiac macrophages compromises repair after myocardial ischemia reperfusion injury. Circ Res 121:930–940. https://doi.org/10.1161/circresaha.117.311327
Deniset JF, Belke D, Lee WY, Jorch SK, Deppermann C, Hassanabad AF, Turnbull JD, Teng G, Rozich I, Hudspeth K, Kanno Y, Brooks SR, Hadjantonakis AK, O'Shea JJ, Weber GF, Fedak PWM, Kubes P (2019) Gata6(+) pericardial cavity macrophages relocate to the injured heart and prevent cardiac fibrosis. Immunity 51:131–140.e135. https://doi.org/10.1016/j.immuni.2019.06.010
Dernellis J, Panaretou M (2004) Relationship between C-reactive protein concentrations during glucocorticoid therapy and recurrent atrial fibrillation. Eur Heart J 25:1100–1107. https://doi.org/10.1016/j.ehj.2004.04.025
Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, Michael LH, Rollins BJ, Entman ML, Frangogiannis NG (2005) CCL2/Monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res 96:881–889. https://doi.org/10.1161/01.RES.0000163017.13772.3a
Dick SA, Macklin JA (2019) Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol 20:29–39. https://doi.org/10.1038/s41590-018-0272-2
Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ, Mann DL (2014) Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40:91–104. https://doi.org/10.1016/j.immuni.2013.11.019
Fayad ZA, Swirski FK, Calcagno C, Robbins CS, Mulder W, Kovacic JC (2018) Monocyte and macrophage dynamics in the cardiovascular system: JACC macrophage in CVD series (Part 3). J Am Coll Cardiol 72:2198–2212. https://doi.org/10.1016/j.jacc.2018.08.2150
Friedrichs K, Klinke A, Baldus S (2011) Inflammatory pathways underlying atrial fibrillation. Trends Mol Med 17:556–563. https://doi.org/10.1016/j.molmed.2011.05.007
Frodermann V, Nahrendorf M (2018) Macrophages and cardiovascular health. Physiol Rev 98:2523–2569. https://doi.org/10.1152/physrev.00068.2017
Fujiu K, Wang J, Nagai R (2014) Cardioprotective function of cardiac macrophages. Cardiovasc Res 102:232–239. https://doi.org/10.1093/cvr/cvu059
Gentek R, Molawi K, Sieweke MH (2014) Tissue macrophage identity and self-renewal. Immunol Rev 262:56–73. https://doi.org/10.1111/imr.12224
Ginhoux F, Guilliams M (2016) Tissue-resident macrophage ontogeny and homeostasis. Immunity 44:439–449. https://doi.org/10.1016/j.immuni.2016.02.024
Ginhoux F, Jung S (2014) Monocytes and macrophages: developmental pathways and tissue homeostasis. Nat Rev Immunol 14:392–404. https://doi.org/10.1038/nri3671
Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, Garner H, Trouillet C, de Bruijn MF, Geissmann F, Rodewald HR (2015) Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 518:547–551. https://doi.org/10.1038/nature13989
Gungor B, Ekmekci A, Arman A, Ozcan KS, Ucer E, Alper AT, Calik N, Yilmaz H, Tezel T, Coker A, Bolca O (2013) Assessment of interleukin-1 gene cluster polymorphisms in lone atrial fibrillation: new insight into the role of inflammation in atrial fibrillation. Pacing Clin Electrophysiol 36:1220–1227. https://doi.org/10.1111/pace.12182
Guo Y, Lip GY, Apostolakis S (2012) Inflammation in atrial fibrillation. J Am Coll Cardiol 60:2263–2270. https://doi.org/10.1016/j.jacc.2012.04.063
Hashimoto D, Chow A, Noizat C, Teo P, Beasley MB, Leboeuf M, Becker CD, See P, Price J, Lucas D, Greter M, Mortha A, Boyer SW, Forsberg EC, Tanaka M, van Rooijen N, Garcia-Sastre A, Stanley ER, Ginhoux F, Frenette PS, Merad M (2013) Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 38:792–804. https://doi.org/10.1016/j.immuni.2013.04.004
Heidt T, Courties G, Dutta P, Sager HB, Sebas M, Iwamoto Y, Sun Y, Da Silva N, Panizzi P, van der Laan AM, Swirski FK, Weissleder R, Nahrendorf M (2014) Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ Res 115:284–295. https://doi.org/10.1161/circresaha.115.303567
Hoeffel G, Chen J, Lavin Y, Low D, Almeida FF, See P, Beaudin AE, Lum J, Low I, Forsberg EC, Poidinger M, Zolezzi F, Larbi A, Ng LG, Chan JK, Greter M, Becher B, Samokhvalov IM, Merad M, Ginhoux F (2015) C-Myb(+) erythro-myeloid progenitor-derived fetal monocytes give rise to adult tissue-resident macrophages. Immunity 42:665–678. https://doi.org/10.1016/j.immuni.2015.03.011
Hoeffel G, Ginhoux F (2018) Fetal monocytes and the origins of tissue-resident macrophages. Cell Immunol 330:5–15. https://doi.org/10.1016/j.cellimm.2018.01.001
Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, Hucker WJ, Wulfers EM, Seemann G, Courties G, Iwamoto Y, Sun Y, Savol AJ, Sager HB, Lavine KJ, Fishbein GA, Capen DE, Da Silva N, Miquerol L, Wakimoto H, Seidman CE, Seidman JG, Sadreyev RI, Naxerova K, Mitchell RN, Brown D, Libby P, Weissleder R, Swirski FK, Kohl P, Vinegoni C, Milan DJ, Ellinor PT, Nahrendorf M (2017) Macrophages Facilitate Electrical Conduction in the Heart. Cell 169:510–522.e520. https://doi.org/10.1016/j.cell.2017.03.050
Jung M, Dodsworth M, Thum T (2018) Inflammatory cells and their non-coding RNAs as targets for treating myocardial infarction. Basic Res Cardiol 114:4. https://doi.org/10.1007/s00395-018-0712-z
Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol 20:4106–4114. https://doi.org/10.1128/mcb.20.11.4106-4114.2000
Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, Ornitz DM, Randolph GJ, Mann DL (2014) Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci USA 111:16029–16034. https://doi.org/10.1073/pnas.1406508111
Lavine KJ, Pinto AR, Epelman S, Kopecky BJ, Clemente-Casares X, Godwin J, Rosenthal N, Kovacic JC (2018) The macrophage in cardiac homeostasis and disease: JACC macrophage in CVD series (Part 4). J Am Coll Cardiol 72:2213–2230. https://doi.org/10.1016/j.jacc.2018.08.2149
Lazzerini PE, Capecchi PL, Laghi-Pasini F (2017) Systemic inflammation and arrhythmic risk: lessons from rheumatoid arthritis. Eur Heart J 38:1717–1727. https://doi.org/10.1093/eurheartj/ehw208
Leid J, Carrelha J, Boukarabila H, Epelman S, Jacobsen SE, Lavine KJ (2016) Primitive Embryonic Macrophages are Required for Coronary Development and Maturation. Circ Res 118:1498–1511. https://doi.org/10.1161/circresaha.115.308270
Leuschner F, Nahrendorf M (2019) Novel functions of macrophages in the heart: insights into electrical conduction, stress, and diastolic dysfunction. Eur Heart J. https://doi.org/10.1093/eurheartj/ehz159
Li W, Hsiao HM, Higashikubo R, Saunders BT, Bharat A, Goldstein DR, Krupnick AS, Gelman AE, Lavine KJ, Kreisel D (2016) Heart-resident CCR2(+) macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight. https://doi.org/10.1172/jci.insight.87315
Mass E, Ballesteros I, Farlik M, Halbritter F, Gunther P, Crozet L, Jacome-Galarza CE, Handler K, Klughammer J, Kobayashi Y, Gomez-Perdiguero E, Schultze JL, Beyer M, Bock C, Geissmann F (2016) Specification of tissue-resident macrophages during organogenesis. Science. https://doi.org/10.1126/science.aaf4238
McGrath KE, Frame JM, Palis J (2015) Early hematopoiesis and macrophage development. Semin Immunol 27:379–387. https://doi.org/10.1016/j.smim.2016.03.013
McGrath KE, Koniski AD, Malik J, Palis J (2003) Circulation is established in a stepwise pattern in the mammalian embryo. Blood 101:1669–1676. https://doi.org/10.1182/blood-2002-08-2531
McGrath KE, Palis J (2005) Hematopoiesis in the yolk sac: more than meets the eye. Exp Hematol 33:1021–1028. https://doi.org/10.1016/j.exphem.2005.06.012
Merz J, Albrecht P, von Garlen S, Ahmed I, Dimanski D, Wolf D, Hilgendorf I, Hardtner C, Grotius K, Willecke F, Heidt T, Bugger H, Hoppe N, Kintscher U, von Zur MC, Idzko M, Bode C, Zirlik A, Stachon P (2018) Purinergic receptor Y2 (P2Y2)- dependent VCAM-1 expression promotes immune cell infiltration in metabolic syndrome. Basic Res Cardiol 113:45. https://doi.org/10.1007/s00395-018-0702-1
Molawi K, Wolf Y, Kandalla PK, Favret J, Hagemeyer N, Frenzel K, Pinto AR, Klapproth K, Henri S, Malissen B, Rodewald HR, Rosenthal NA, Bajenoff M, Prinz M, Jung S, Sieweke MH (2014) Progressive replacement of embryo-derived cardiac macrophages with age. J Exp Med 211:2151–2158. https://doi.org/10.1084/jem.20140639
Moore KJ, Koplev S, Fisher EA, Tabas I, Bjorkegren JLM, Doran AC, Kovacic JC (2018) Macrophage trafficking, inflammatory resolution, and genomics in atherosclerosis: JACC macrophage in CVD series (Part 2). J Am Coll Cardiol 72:2181–2197. https://doi.org/10.1016/j.jacc.2018.08.2147
Moore MA, Metcalf D (1970) Ontogeny of the haemopoietic system: yolk sac origin of in vivo and in vitro colony forming cells in the developing mouse embryo. Br J Haematol 18:279–296. https://doi.org/10.1111/j.1365-2141.1970.tb01443.x
Mouton AJ, DeLeon-Pennell KY, Rivera Gonzalez OJ, Flynn ER, Freeman TC, Saucerman JJ, Garrett MR, Ma Y, Harmancey R, Lindsey ML (2018) Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res Cardiol 113:26. https://doi.org/10.1007/s00395-018-0686-x
Mylonas KJ, Jenkins SJ, Castellan RF, Ruckerl D, McGregor K, Phythian-Adams AT, Hewitson JP, Campbell SM, MacDonald AS, Allen JE, Gray GA (2015) The adult murine heart has a sparse, phagocytically active macrophage population that expands through monocyte recruitment and adopts an 'M2' phenotype in response to Th2 immunologic challenge. Immunobiology 220:924–933. https://doi.org/10.1016/j.imbio.2015.01.013
Nahrendorf M, Swirski FK (2013) Monocyte and macrophage heterogeneity in the heart. Circ Res 112:1624–1633. https://doi.org/10.1161/circresaha.113.300890
Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, Libby P, Weissleder R, Pittet MJ (2007) The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med 204:3037–3047. https://doi.org/10.1084/jem.20070885
Naito M, Yamamura F, Nishikawa S, Takahashi K (1989) Development, differentiation, and maturation of fetal mouse yolk sac macrophages in cultures. J Leukoc Biol 46:1–10. https://doi.org/10.1002/jlb.46.1.1
Patel P, Dokainish H, Tsai P, Lakkis N (2010) Update on the association of inflammation and atrial fibrillation. J Cardiovasc Electrophysiol 21:1064–1070. https://doi.org/10.1111/j.1540-8167.2010.01774.x
Pinto AR, Godwin JW, Chandran A, Hersey L, Ilinykh A, Debuque R, Wang L, Rosenthal NA (2014) Age-related changes in tissue macrophages precede cardiac functional impairment. Aging (Albany NY) 6:399–413. https://doi.org/10.18632/aging.100669
Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D'Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD (2016) Revisiting Cardiac Cellular Composition. Circ Res 118:400–409. https://doi.org/10.1161/circresaha.115.307778
Pinto AR, Paolicelli R, Salimova E, Gospocic J, Slonimsky E, Bilbao-Cortes D, Godwin JW, Rosenthal NA (2012) An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS ONE 7:e36814. https://doi.org/10.1371/journal.pone.0036814
Podaru MN, Fields L, Kainuma S, Ichihara Y, Hussain M, Ito T, Kobayashi K, Mathur A, D'Acquisto F, Lewis-McDougall F, Suzuki K (2019) Reparative macrophage transplantation for myocardial repair: a refinement of bone marrow mononuclear cell-based therapy. Basic Res Cardiol 114:34. https://doi.org/10.1007/s00395-019-0742-1
Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, Gorbatov R, Sukhova GK, Gerhardt LM, Smyth D, Zavitz CC, Shikatani EA, Parsons M, van Rooijen N, Lin HY, Husain M, Libby P, Nahrendorf M, Weissleder R, Swirski FK (2013) Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med 19:1166–1172. https://doi.org/10.1038/nm.3258
Roth GA, Abate D, Abate KH, Abay SM, Abbafati C, Abbasi N, Abbastabar H, Abd-Allah L, Abdela J, Abdelalim A, Abdollahpour I, Abdulkader RS, Abebe HT, Abebe M, Abebe Z, Abejie AN, Abera SF, Abil OZ, Abraha HN, Abrham AR, Abu-Raddad LJ, Accrombessi MMK, Acharya D, Adamu AA, Adebayo O, Adedoyin RA, Adekanmbi V, Adookunboh O, Adhena BM, Adib MG, Admasie A, Afshin A, Agarwal G, Agesa KM, Agrawal A, Agrawal S, Ahmadi A, Ahmadi M, Ahmed MB, Ahmed S, Aichour AN, Aichour I, Aichour MTF, Akbari ME, Akinyeniti RO, Akseer N, Al-Aly Z, Al-Eyadhy A, Al-Raddadi RM, Alandab F, Alam K, Alam T, Alebel A, Alene KA, Alijanzadeh M, Alizadeh-Navaei R, Aljunid SM, Aa A, Alla F, Allebeck P, Alonso J, Altirkawi K, Alvis-Guzman N, Amare AT, Aminde LN, Amini E, Ammar W, Amoako YA, Anber NH, Andrei CL, Androudi S, Animut MD, Anjomshoa M, Ansari H, Aniha MG, Antonio CAT, Anwari P, Aremu O, Arnlov J, Arora A, Arora M, Artaman A, Aryal KK, Asayesh H, Asfaw ET, Ataro Z, Atique S, Atre SR, Ausloos M, Avokpaho EFGA, Awasthi A, Quintattilla BPA, Ayele Y, Ayer R, Azzopardi PS, Babazadeh A, Bacha U, Badali H, Badawi A, Bali AG et al (2018) Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet 392:1736–1788. https://doi.org/10.1016/s0140-6736(18)32203-7
Samokhvalov IM, Samokhvalova NI, Nishikawa S (2007) Cell tracing shows the contribution of the yolk sac to adult haematopoiesis. Nature 446:1056–1061. https://doi.org/10.1038/nature05725
Schulz C, Gomez Perdiguero E, Chorro L, Szabo-Rogers H, Cagnard N, Kierdorf K, Prinz M, Wu B, Jacobsen SE, Pollard JW, Frampton J, Liu KJ, Geissmann F (2012) A lineage of myeloid cells independent of Myb and hematopoietic stem cells. Science 336:86–90. https://doi.org/10.1126/science.1219179
Stevens SM, von Gise A, VanDusen N, Zhou B, Pu WT (2016) Epicardium is required for cardiac seeding by yolk sac macrophages, precursors of resident macrophages of the adult heart. Dev Biol 413:153–159. https://doi.org/10.1016/j.ydbio.2016.03.014
Sun Z, Zhou D, Xie X, Wang S, Wang Z, Zhao W, Xu H, Zheng L (2016) Cross-talk between macrophages and atrial myocytes in atrial fibrillation. Basic Res Cardiol 111:63. https://doi.org/10.1007/s00395-016-0584-z
Takahashi K, Naito M (1993) Development, differentiation, and proliferation of macrophages in the rat yolk sac. Tissue Cell 25:351–362. https://doi.org/10.1016/0040-8166(93)90077-x
Takahashi K, Yamamura F, Naito M (1989) Differentiation, maturation, and proliferation of macrophages in the mouse yolk sac: a light-microscopic, enzyme-cytochemical, immunohistochemical, and ultrastructural study. J Leukoc Biol 45:87–96. https://doi.org/10.1002/jlb.45.2.87
Vagnozzi RJ, Maillet M, Sargent MA, Khalil H, Johansen AK, Schwanekamp JA, York AJ, Huang V, Nahrendorf M, Sadayappan S, Molkentin JD (2019) An acute immune response underlies the benefit of cardiac stem-cell therapy. Nature. https://doi.org/10.1038/s41586-019-1802-2
van Furth R, Cohn ZA (1968) The origin and kinetics of mononuclear phagocytes. J Exp Med 128:415–435. https://doi.org/10.1084/jem.128.3.415
Wang Z, Cui M, Shah AM, Ye W, Tan W, Min YL, Botten GA, Shelton JM, Liu N, Bassel-Duby R, Olson EN (2019) Mechanistic basis of neonatal heart regeneration revealed by transcriptome and histone modification profiling. Proc Natl Acad Sci USA 116:18455–18465. https://doi.org/10.1073/pnas.1905824116
Wang Z, Koenig AL, Lavine KJ, Apte RS (2019) Macrophage Plasticity and Function in the Eye and Heart. Trends Immunol 40:825–841. https://doi.org/10.1016/j.it.2019.07.002
Williams JW, Giannarelli C, Rahman A, Randolph GJ, Kovacic JC (2018) Macrophage biology, classification, and phenotype in cardiovascular disease: JACC macrophage in CVD series (Part 1). J Am Coll Cardiol 72:2166–2180. https://doi.org/10.1016/j.jacc.2018.08.2148
Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38:79–91. https://doi.org/10.1016/j.immuni.2012.12.001
Zeng Y, He J, Bai Z (2019) Tracing the first hematopoietic stem cell generation in human embryo by single-cell RNA sequencing. Cell Res. https://doi.org/10.1038/s41422-019-0228-6
Funding
This work was supported by the Nature Science Foundation of Zhejiang Province (Grant no. LZ16H020001); the National Key R&D Program of China (Grant no. 2016YFC1301003); the National Science Foundation of China (Grant no. 81873484); the Natural Science Foundation of Zhejiang Province, Zhejiang, China, Grant/Award Number: LQ19H070002.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There are no conflicts of interests to be declared by all authors.
Rights and permissions
About this article

Cite this article
Wang, Z., Lu, YL., Zhao, WT. et al. Distinct origins and functions of cardiac orthotopic macrophages. Basic Res Cardiol 115, 8 (2020). https://doi.org/10.1007/s00395-019-0769-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-019-0769-3