
Abstract
Purpose of Review
The cardiac immune landscape dynamically changes in response to aging, hemodynamic stress, and myocardial injury. Here, we highlight key cardiac immune cell types, their role in reshaping the cellular landscape and promoting tissue remodeling following cardiac insults, and how understanding of these processes uncovers novel disease mechanisms that contribute to cardiac pathology.
Recent Findings
Distinct subsets of cardiac macrophages reside within the heart and exhibit divergent functions in response to myocardial injury. Parsing cardiac macrophages based on developmental origin has served as a valuable approach to define functionally divergent populations of reparative (embryonic-derived, tissue resident) and inflammatory (monocyte-derived, recruited) cardiac macrophages. Single-cell transcriptomics and elucidation of the effector mechanisms that orchestrate macrophage functions has provided new and therapeutically tractable insights into the pathogenesis of numerous cardiac diseases.
Summary
The immune landscape of the heart is dynamic and represents an important mediator of disease pathogenesis across an array of cardiac pathology. Elucidation of mechanisms that drive inflammatory monocyte/macrophage recruitment, activation, and effector responses may lead to the identification of new therapeutic targets.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The role of the immune system during cardiac homeostasis, aging, and disease is an active area of investigation. In the steady state, the mouse and human heart is comprised of cardiomyocytes, fibroblasts, endothelial cells, smooth muscle cells, pericytes, endocardial cells, epicardial cells, and various immune cells including monocytes, macrophages, dendritic cells, T-cells, natural killer (NK)-cells, mast cells, and B-cells [1, 2, 3•]. Among these immune populations, macrophages represent the most prevalent cell type. The cardiac immune landscape is dynamic and undergoes dramatic composition and phenotype shifts that differ based on the disease context (i.e., infection, sterile injury, hemodynamic perturbations, aging, myocarditis). In this review, we will discuss the cardiac inflammatory response following acute myocardial injury.
Composition of the Cardiac Immune Landscape
The cardiac immune landscape dynamically changes as it transitions from homeostasis and surveillance to an inflammatory state. In the steady state, the heart contains immune cells that are interspersed within cardiac tissue. The bulk of these immune cells are tissue-resident macrophages [4]. As part of the innate immune system, macrophages are professional phagocytes with various capabilities including recognizing and destroying apoptotic cells and pathogens as well as communicating with surrounding stromal and immune cells [5].
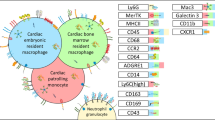
Identification and classification of tissue-resident cardiac macrophage subtypes has been achieved through the use of cell surface markers (CCR2: chemokine receptor-2; LYVE1: lymphatic vessel endothelial hyaluronan receptor 1; and TIMD4: T-cell immunoglobulin and mucin domain containing 4), genetic fate mapping, transcriptomic profiling, and functional analysis [6,7,8,9,10, 11•, 12]. In the steady state, CCR2−LYVE1+TIMD4+ tissue-resident macrophages comprise the bulk of all immune cells within the heart (Fig. 1). Cardiac tissue-resident macrophages (CRMs) are long-lived, originate from embryonic hematopoietic progenitors that arise within the yolk sac, and seed the heart during embryonic development [6, 7, 9, 10, 13, 14]. CRMs are maintained independent of monocyte input through local proliferation within the heart.

Distribution and key features of macrophage subsets in the heart. The heart primarily contains chemokine receptor-2 (CCR2−) tissue-resident macrophages in the steady state, during neonatal heart regeneration, and in adoptive remodeling. There is a shift towards CCR2+ macrophage expansion in sterile injury, myocarditis, chronic heart failure, and aging. Key features of each subset are highlighted in the colored corresponding boxes. AngII/PE, angiotensin II and phenylephrine (created with BioRender.com)
Multiple studies have demonstrated that CRMs have interesting and unique tissue specific functions, similar to what has been observed for other tissue-resident macrophage populations (i.e., microglia, Kuepfer cells, alveolar macrophages) [13, 15,16,17,18]. CRMs are active during development and homeostasis, where they function to clear apoptotic cells, promote coronary growth and patterning, clear damaged mitochondria, and facilitate electrical conduction [8, 11•, 19, 20]. CRMs suppress inflammation by preventing monocyte and neutrophil recruitment potentially through secretion of interleukin (IL)-10 and/or transforming growth factor (TGF)-β [21]. CRMs orchestrate repair and regeneration of the neonatal heart by regulating myocardial proliferation and angiogenesis [9, 22, 23]. In the context of chronic heart failure, CRMs are activated by mechanical stretch through transient receptor potential cation channel subfamily V member 4 (TRPV4) and trigger adaptations necessary to maintain cardiac output including reorganization of myocardial tissue and coronary angiogenesis [24••, 25, 26]. CRMs that express high levels of major histocompatibility complex (MHC)-II also have the capacity to present antigens; however, the functional relevance of this property is not known [8].
Under homeostatic conditions, CRMs are admixed with CCR2+MHC-IIhi macrophages, which are derived from monocytes (Fig. 1). CCR2+MHC-IIhi macrophages seed the heart postnatally (~ 2 weeks after birth) and are maintained through ongoing monocyte recruitment and subsequent proliferation. CCR2+MHC-IIhi macrophages have robust inflammatory potential and secrete chemokines that support neutrophil and monocyte recruitment [6,7,8,9, 14]. The heart also contains a small number of lymphocyte antigen 6 complex (Ly6C)hiCCR2+ monocytes that can be distinguished from cardiac macrophages based on the expression of Mer proto-oncogene tyrosine kinase (MertK) [9, 27].
Lastly, the heart contains small numbers of dendritic cells, mast cells, memory T-cells, regulatory T-cells, and intravascular B-cells that contribute to immunosurveillance in the steady state. In this scenario, dendritic cells are thought to serve as gatekeepers of self-tolerance [28, 29]. The exact role of these populations remains to be fully elucidated.
Immune Response to Myocardial Injury
A diverse array of insults including ischemia, hypertension, autoimmunity, genetic mutations, chemical toxicity, and infection may lead to cardiac injury, cell death, and resultant inflammation. We define necroinflammation as the immune response to necrosis in a living organism. The coupling of cell death to inflammation and the immune systems response is referred to as necroinflammation [30]. Necrosis and programmed forms of necrotic cell death (ferroptosis, necroptosis, pyroptosis) promote release of intracellular materials that are recognized by immune cells and initiate inflammation. Collectively, these substances are referred to as alarmins, damage-associated molecular patterns (DAMPs), or pathogen-associated molecular patterns (PAMPs) depending on whether they are released by host cells or pathogens [31, 32] (Fig. 2). DAMPs and PAMPs consist of lipids, proteins, carbohydrates, or nucleic acids and are recognized by pattern recognition receptors (PRRs), such as Toll-like receptors (TLRs).

Cardiac immune cells at steady state, following cardiac injury, and post-injury. In the steady state, the heart primarily contains chemokine receptor-2 (CCR2−) tissue-resident macrophages (CRMs) with few other immune cells including dendritic cells and T-cells. Activation of the cardiac immune response following cardiac injury begins with innate immune cells binding damage-associated molecular patterns (DAMPs) through pattern recognition receptors (PRRs). Pro-inflammatory cytokines and chemokines are released into the bloodstream, leading to recruitment and activation of CCR2+ major histocompatibility complex (MHC)-IIhi, monocytes, and neutrophils, which then triggers activation of the adaptive immune system by recruiting B-cells and T-cells to the damaged myocardium. Additional immune cells such as mast cells, eosinophils, and natural killer (NK) cells may also be recruited to the site of injury. Finally, in a process that is dependent on CRMs, the post-injury heart undergoes remodeling through myofibroblast-generated extracellular matrix that is required for heart repair (created with BioRender.com)
Within the heart, ferroptosis and necroptosis have been implicated as major forms of cardiomyocyte cell death [33]. There is increasing recognition that cardiomyocyte ferroptosis is a common form of cell death and is causally linked with myocardial inflammation and neutrophil recruitment [34]. The exact mediators (alarmins, DAMPs) released by ferroptotic cardiomyocytes remain an open area of investigation. Regardless, TLR4 and TLR9 appear to serve as PRRs, which are expressed on CRMs and CCR2+MHC-IIhi macrophages [34,35,36]. TLR signaling in CCR2+MHC-IIhi macrophages triggers downstream gene expression of pro-inflammatory cytokines such as IL-1β, IL-6, and tumor necrosis factor (TNF) as well as chemokines such as CC-chemokine ligand 2 (CCL2), CCL3, CCL5, and CCL17 that are associated with negative inotropic effects, left ventricular (LV) dysfunction, and LV remodeling [37,38,39,40,41,42]. Cytokines and chemokines directly activate endothelial cells, recruit circulating monocytes and neutrophils from the bloodstream, and direct their extravasation into the myocardium. The role of TLR signaling in CRMs remains to be defined.
Acute myocardial injury triggers a dramatic shift in the cardiac immune landscape (Fig. 2). Loss of CRMs in conjunction with infiltration of neutrophils, monocytes, monocyte-derived macrophages, and dendritic cells has been observed in response to multiple forms of cardiac injury including permanent myocardial infarction, reperfused myocardial infarction, diphtheria toxin-mediated cardiomyocyte ablation, transverse aortic constriction, angiotensin II and phenylephrine infusion, and myocarditis [8, 9, 43,44,45,46,47,48]. Neutrophil infiltration and monocyte recruitment are initiated by CCR2+MHC-IIhi macrophages within the heart as depletion of this population results in marked reductions in neutrophil extravasation and monocyte recruitment [6, 36]. Following ischemia reperfusion injury, CCR2+MHC-IIhi macrophages generate and secrete multiple chemokines (CCL2, CCL7, CXCL2, CXCL5) and cytokines (TNF, IL-1β) important for peripheral leukocyte recruitment. Each of these factors is dependent on myeloid differentiation primary response 88 (MYD88) signaling, an important mediator of TLR activation. CCR2+MHC-IIhi macrophages also generate and secrete CCL17, a pro-inflammatory factor that drives inflammation, myocardial fibrosis, and LV remodeling. CCL17 expression is triggered by granulocyte–macrophage colony-stimulating factor (GM-CSF) signaling and functions to prevent regulatory T-cell recruitment, highlighting the importance of crosstalk between macrophages and T-cells [42].
Intriguingly, CRMs appear to suppress neutrophil and monocyte recruitment through a mechanism that remains to be defined. In addition to their anti-inflammatory effects, CRMs also regulate lymphangiogenesis, a process that may be important for the resolution of inflammation [49]. Importantly, selective depletion of CCR2+MHC-IIhi macrophages or CRMs results in divergent effects on LV systolic function and cardiac remodeling following tissue injury highlighting the significance of cardiac macrophages in disease [6, 24••].
Plasticity and Diversification of Monocytes and Monocyte-Derived Cells
Monocytes display extraordinary plasticity and may differentiate into a variety of cell types [8, 15,16,17]. Recent advances in single-cell RNA sequencing technologies have made it possible to resolve the myriad of cell states and phenotypes acquired by monocytes as they enter and expand within the injured myocardium. Following myocardial infarction, at least 7 morphologically and transcriptionally distinct subsets of monocytes, macrophages, and dendritic cells have been identified within the myocardium [6, 11•, 50]. The majority of these cell types are unique to the injured heart, highlighting how the cardiac immune landscape is reshaped by injury and disease. The exact function of these populations and mechanisms responsible for their differentiation and longevity is under active investigation. It is likely that monocyte fate decisions are heavily influenced by cues derived from their local environment. For example, monocytes that enter the infarct and peri-infarct zones are exposed to differing environments such as tissue hypoxia and oxidative intermediates, respectively [11•, 51]. Consistent with this premise, arginase 1 (Arg1)+ macrophages are located within the infarct, while interferon-activated macrophages are found within the border zone [6].
Flow cytometry, immunostaining, and single-cell RNA sequencing studies have uncovered that the human heart harbors monocyte and macrophage populations that are functionally analogous to those found in the mouse heart. Under steady state conditions, the human heart contains CCR2−LYVE1+ CRMs and CCR2+MHC-IIhi macrophages with transcriptional profiles and inflammatory potential that resemble their mouse counterparts [7]. The abundance of CCR2+MHC-IIhi macrophages is associated with poor outcomes including persistent LV dysfunction following mechanical unloading. More recently, single-cell RNA sequencing of healthy human and heart failure tissue has revealed that the immune landscape of the human heart is also reshaped by disease. Chronic heart failure was associated with loss of CRMs and emergence of several monocyte and monocyte-derived macrophage populations [3•].
Aging
Inflammaging is a phenomenon characterized by upregulation of the basal inflammatory response that is associated with aging [52]. As such, the cardiac immune landscape changes with aging, which is most notable with a decrease in CRM replication and a concomitant increase in pro-inflammatory CCR2+MHC-IIhi macrophages [53]. In addition to cardiac macrophage population changes, there is an increase in basal circulating pro-inflammatory cytokines (TNF-α) and chemokines (CCL2), reflective of dysregulation of the innate immune system associated with aging [54, 55]. Research in clonal hematopoiesis reflects the recognition of the intersection between inflammaging and the expansion of mutated hematopoietic stem cell clones, reshaping the cardiac immune landscape with mutated pro-inflammatory myeloid cells [56]. In a mouse model of clonal hematopoiesis, ten-eleven translocation 2–mutant (Tet2)-deficient mice demonstrated cardiac expansion of CCR2+MHC-IIhi macrophages but not CRMs, which exhibited an inflammatory transcriptome profile in conjunction with age-related cardiac dysfunction [57].
In line with inflammaging, cardiac immunosenescence is the process by which the immune system deteriorates with aging. Macrophages have impaired phagocytic function as well as decreased polarization (pro- or anti-inflammatory) when exposed to pathogens [58, 59]. In addition, studies have shown impaired TLR expression and function in innate immune cells [60]. Collectively, inflammaging, clonal hematopoiesis, and immunosenescence within cardiac immune cells contribute to the notion that age is a risk factor in cardiovascular disease. The precise cell states associated with aging are incompletely understood and a topic of ongoing interest. Single-cell omic studies would be highly informative to clarify if cell states associated with aging are unique or reminiscent of those seen in acute or chronic inflammation.
Non-invasive Imaging of the Cardiac Immune Landscape
A key limitation of studying human cardiac immune cells is the reliance on myocardial specimens and biases imposed by selectively studying individuals undergoing endomyocardial biopsy, left ventricular assist device implantation, and/or heart transplantation. Molecular imaging offers a non-invasive solution to investigate cardiac immune cell composition, the natural history of inflammation, and establish associations between particular immune subsets and relevant patient outcomes. We have developed a positron emission tomography radiotracer that allosterically binds to CCR2, 68 Ga-DOTA [1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid]-ECL1i [extracellular loop 1 inverso]. 68 Ga-DOTA-ECL1i PET/CT readily visualizes CCR2+ monocytes and macrophages in various cardiac injury mouse models [61]. Recently, we have translated this tracer for human investigation in pulmonary fibrosis [62]. We are currently assessing its utility in imaging patients that suffered an acute myocardial infarction. We envision that these studies will provide key information pertaining to the natural history of CCR2+ monocyte and macrophage accumulation following human myocardial infarction and identify imaging features associated with clinical outcomes. Similar strategies are being developed to image activated fibroblast (FAPI-46), neutrophils, T-cells, and CRMs [63, 64].
New Frontiers of Cardio-immunology
As the cardiac immune landscape continues to be defined within the confines of established cardiac injury models (myocardial infarction, pressure overload, viral myocarditis), it is imperative to also consider emerging disease entities with profound effects on survival and quality of life. For instance, immunotherapy continues to revolutionize the oncology field as it holds the potential to control and possibly cure previously untreatable cancers. However, immune checkpoint inhibitors, such as programmed cell death protein 1 (PD-1), programmed death-ligand 1 (PD-L1), and cytotoxic T-lymphocyte associated protein 4 (CTLA-4), are associated with myocarditis [65, 66]. Established immune checkpoint inhibitors function by blocking inhibitory receptor-ligand interactions to augment T-cell-mediated killing of cancer cells. Based on the success of these agents, there is an explosion of new immune checkpoint therapeutics, such as cluster of differentiation (CD40) and CD134 (also known as OX40) agonist antibodies and lymphocyte activating 3 (Lag3) neutralizing antibodies [67,68,69,70,71]. These therapies are expected to further enhance T-cell responses and augment activation of antigen presenting cells (macrophages, dendritic cells). The potential for immune checkpoint therapies to reshape the cardiac immune landscape is clear. Understanding the impact of checkpoint therapies on cardiac immune cell composition and function is needed to understand short-term and long-term toxicities as well as ramification on cardiac homeostasis and disease progression.
Recent work has established a mouse model that recapitulates checkpoint inhibitor-associated myocarditis, which demonstrated macrophage and T-cell myocardial infiltration that was associated with premature death [45]. It is unclear how immune checkpoint inhibitors promote macrophage and T-cell expansion and how these immune populations might differ from those found in the infarcted heart. In addition, the relevant contribution of T-cells and macrophages to myocarditis and the precise effector molecules involved remain to be defined. A detailed understanding of these mechanisms will be required to adequately treat this devastating disease without adversely affecting anti-tumor responses and long-term tumor control. Finally, little is understood regarding the chronic effects of immune checkpoint inhibitors on cardiac homeostasis, function, and aging. These examples highlight the clinical utility and importance of the growing field of cardio-immunology.
Conclusion
Cardiac injury activates a complex inflammatory response comprised of the innate and adaptive immune systems. In the heart, CRMs are the most abundant immune cell in the steady state. However, following myocardial injury, the immune landscape shifts as a result of monocyte infiltration, diversification, and expansion of pro-inflammatory macrophage populations including CCR2+MHC-IIhi macrophages. Single-cell technologies and downstream functional studies have allowed tremendous progress in defining the immune landscape of the heart and identifying therapeutic targets. Understanding the mechanistic basis for necroinflammation, monocyte infiltration, and fate decisions necessary for the specification of inflammatory populations is crucial to developing new therapies that may improve patient outcomes.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, et al. Revisiting cardiac cellular composition. Circ Res. 2016;118(3):400–9. https://doi.org/10.1161/CIRCRESAHA.115.307778.
Tucker NR, Chaffin M, Fleming SJ, Hall AW, Parsons VA, Bedi KC Jr, et al. Transcriptional and cellular diversity of the human heart. Circulation. 2020;142(5):466–82. https://doi.org/10.1161/CIRCULATIONAHA.119.045401.
• Koenig AL, Shchukina I, Amrute J, Andhey PS, Zaitsev K, Lai L, et al. Single-cell transcriptomics reveals cell-type-specific diversification in human heart failure. Nat Cardiovasc Res. 2022;1(3):263–80. https://doi.org/10.1038/s44161-022-00028-6. Findings provide a comprehensive analysis of the cellular and transcriptomic landscape of human heart failure, identify cell type-specific transcriptional programs and disease-associated cell states and establish a valuable resource for the investigation of human heart failure.
Ramos GC, van den Berg A, Nunes-Silva V, Weirather J, Peters L, Burkard M, et al. Myocardial aging as a T-cell-mediated phenomenon. Proc Natl Acad Sci U S A. 2017;114(12):E2420–9. https://doi.org/10.1073/pnas.1621047114.
Metchnikoff E. Immunity in infective diseases, F.G. Binnie (translation). Cambridge University Press, London. 1905.
Bajpai G, Bredemeyer A, Li W, Zaitsev K, Koenig AL, Lokshina I, et al. Tissue resident CCR2- and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circ Res. 2019;124(2):263–78. https://doi.org/10.1161/CIRCRESAHA.118.314028.
Bajpai G, Schneider C, Wong N, Bredemeyer A, Hulsmans M, Nahrendorf M, et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat Med. 2018;24(8):1234–45. https://doi.org/10.1038/s41591-018-0059-x.
Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity. 2014;40(1):91–104. https://doi.org/10.1016/j.immuni.2013.11.019.
Lavine KJ, Epelman S, Uchida K, Weber KJ, Nichols CG, Schilling JD, et al. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc Natl Acad Sci U S A. 2014;111(45):16029–34. https://doi.org/10.1073/pnas.1406508111.
Leid J, Carrelha J, Boukarabila H, Epelman S, Jacobsen SE, Lavine KJ. Primitive embryonic macrophages are required for coronary development and maturation. Circ Res. 2016;118(10):1498–511. https://doi.org/10.1161/CIRCRESAHA.115.308270.
• Dick SA, Macklin JA, Nejat S, Momen A, Clemente-Casares X, Althagafi MG, et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat Immunol. 2019;20(1):29–39. https://doi.org/10.1038/s41590-018-0272-2. Findings demonstrate four populations of cardiac macrophages, including a tissue resident subset that is maintained independently of blood monocytes. These macrophage subsets diversify following ischemic injury, most notably with a marked reduction in resident macrophage abundance within the infarct zone that demonstrates a nonredundant, cardioprotective role of tissue resident cardiac macrophages.
Chakarov S, Lim HY, Tan L, Lim SY, See P, Lum J, et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science. 2019;363(6432). https://doi.org/10.1126/science.aau0964.
Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518(7540):547–51. https://doi.org/10.1038/nature13989.
Nahrendorf M, Swirski FK, Aikawa E, Stangenberg L, Wurdinger T, Figueiredo JL, et al. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J Exp Med. 2007;204(12):3037–47. https://doi.org/10.1084/jem.20070885.
Guilliams M, De Kleer I, Henri S, Post S, Vanhoutte L, De Prijck S, et al. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via GM-CSF. J Exp Med. 2013;210(10):1977–92. https://doi.org/10.1084/jem.20131199.
Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 2010;330(6005):841–5. https://doi.org/10.1126/science.1194637.
Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013;38(1):79–91. https://doi.org/10.1016/j.immuni.2012.12.001.
Crofton RW, Diesselhoff-den Dulk MM, van Furth R. The origin, kinetics, and characteristics of the Kupffer cells in the normal steady state. J Exp Med. 1978;148(1):1–17. https://doi.org/10.1084/jem.148.1.1.
Hulsmans M, Clauss S, Xiao L, Aguirre AD, King KR, Hanley A, et al. Macrophages facilitate electrical conduction in the heart. Cell. 2017;169(3):510–22 e20. https://doi.org/10.1016/j.cell.2017.03.050.
Nicolas-Avila JA, Lechuga-Vieco AV, Esteban-Martinez L, Sanchez-Diaz M, Diaz-Garcia E, Santiago DJ, et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell. 2020;183(1):94–109 e23. https://doi.org/10.1016/j.cell.2020.08.031.
Hilgendorf I, Gerhardt LM, Tan TC, Winter C, Holderried TA, Chousterman BG, et al. Ly-6Chigh monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ Res. 2014;114(10):1611–22. https://doi.org/10.1161/CIRCRESAHA.114.303204.
Aurora AB, Porrello ER, Tan W, Mahmoud AI, Hill JA, Bassel-Duby R, et al. Macrophages are required for neonatal heart regeneration. J Clin Invest. 2014;124(3):1382–92. https://doi.org/10.1172/JCI72181.
Wang Z, Cui M, Shah AM, Ye W, Tan W, Min YL, et al. Mechanistic basis of neonatal heart regeneration revealed by transcriptome and histone modification profiling. Proc Natl Acad Sci U S A. 2019;116(37):18455–65. https://doi.org/10.1073/pnas.1905824116.
•• Wong NR, Mohan J, Kopecky BJ, Guo S, Du L, Leid J, et al. Resident cardiac macrophages mediate adaptive myocardial remodeling. Immunity. 2021;54(9):2072–88 e7. https://doi.org/10.1016/j.immuni.2021.07.003. Findings establish a role for tissue-resident macrophages in adaptive cardiac remodeling and implicate mechanical sensing through TRPV-4 in cardiac macrophage activation.
Zaman R, Hamidzada H, Kantores C, Wong A, Dick SA, Wang Y, et al. Selective loss of resident macrophage-derived insulin-like growth factor-1 abolishes adaptive cardiac growth to stress. Immunity. 2021;54(9):2057–71 e6. https://doi.org/10.1016/j.immuni.2021.07.006.
Revelo XS, Parthiban P, Chen C, Barrow F, Fredrickson G, Wang H, et al. Cardiac resident macrophages prevent fibrosis and stimulate angiogenesis. Circ Res. 2021;129(12):1086–101. https://doi.org/10.1161/CIRCRESAHA.121.319737.
Gautier EL, Shay T, Miller J, Greter M, Jakubzick C, Ivanov S, et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat Immunol. 2012;13(11):1118–28. https://doi.org/10.1038/ni.2419.
Hart DN, Fabre JW. Demonstration and characterization of Ia-positive dendritic cells in the interstitial connective tissues of rat heart and other tissues, but not brain. J Exp Med. 1981;154(2):347–61. https://doi.org/10.1084/jem.154.2.347.
Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, et al. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med. 2012;209(6):1135–52. https://doi.org/10.1084/jem.20120030.
Sarhan M, Land WG, Tonnus W, Hugo CP, Linkermann A. Origin and consequences of necroinflammation. Physiol Rev. 2018;98(2):727–80. https://doi.org/10.1152/physrev.00041.2016.
Mackey D, McFall AJ. MAMPs and MIMPs: proposed classifications for inducers of innate immunity. Mol Microbiol. 2006;61(6):1365–71. https://doi.org/10.1111/j.1365-2958.2006.05311.x.
Rock KL, Latz E, Ontiveros F, Kono H. The sterile inflammatory response. Annu Rev Immunol. 2010;28:321–42. https://doi.org/10.1146/annurev-immunol-030409-101311.
Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. 2019;99(4):1765–817. https://doi.org/10.1152/physrev.00022.2018.
Li W, Feng G, Gauthier JM, Lokshina I, Higashikubo R, Evans S, et al. Ferroptotic cell death and TLR4/Trif signaling initiate neutrophil recruitment after heart transplantation. J Clin Invest. 2019;129(6):2293–304. https://doi.org/10.1172/JCI126428.
Mann DL. The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls. Circ Res. 2011;108(9):1133–45. https://doi.org/10.1161/CIRCRESAHA.110.226936.
Li W, Hsiao HM, Higashikubo R, Saunders BT, Bharat A, Goldstein DR, et al. Heart-resident CCR2(+) macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight. 2016;1(12). https://doi.org/10.1172/jci.insight.87315.
Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5(10):987–95. https://doi.org/10.1038/ni1112.
Bozkurt B, Kribbs SB, Clubb FJ Jr, Michael LH, Didenko VV, Hornsby PJ, et al. Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation. 1998;97(14):1382–91. https://doi.org/10.1161/01.cir.97.14.1382.
Gulick T, Chung MK, Pieper SJ, Lange LG, Schreiner GF. Interleukin 1 and tumor necrosis factor inhibit cardiac myocyte beta-adrenergic responsiveness. Proc Natl Acad Sci U S A. 1989;86(17):6753–7. https://doi.org/10.1073/pnas.86.17.6753.
Yokoyama T, Vaca L, Rossen RD, Durante W, Hazarika P, Mann DL. Cellular basis for the negative inotropic effects of tumor necrosis factor-alpha in the adult mammalian heart. J Clin Invest. 1993;92(5):2303–12. https://doi.org/10.1172/JCI116834.
Kakio T, Matsumori A, Ono K, Ito H, Matsushima K, Sasayama S. Roles and relationship of macrophages and monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 in the ischemic and reperfused rat heart. Lab Invest. 2000;80(7):1127–36. https://doi.org/10.1038/labinvest.3780119.
Feng G, Bajpai G, Ma P, Koenig A, Bredemeyer A, Lokshina I, et al. CCL17 aggravates myocardial injury by suppressing recruitment of regulatory T cells. Circulation. 2022;145(10):765–82. https://doi.org/10.1161/CIRCULATIONAHA.121.055888.
Liao X, Shen Y, Zhang R, Sugi K, Vasudevan NT, Alaiti MA, et al. Distinct roles of resident and nonresident macrophages in nonischemic cardiomyopathy. Proc Natl Acad Sci U S A. 2018;115(20):E4661–9. https://doi.org/10.1073/pnas.1720065115.
Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M, et al. CCR2(+) Monocyte-derived infiltrating macrophages are required for adverse cardiac remodeling during pressure overload. JACC Basic Transl Sci. 2018;3(2):230–44. https://doi.org/10.1016/j.jacbts.2017.12.006.
Wei SC, Meijers WC, Axelrod ML, Anang NAS, Screever EM, Wescott EC, et al. A genetic mouse model recapitulates immune checkpoint inhibitor-associated myocarditis and supports a mechanism-based therapeutic intervention. Cancer Discov. 2021;11(3):614–25. https://doi.org/10.1158/2159-8290.CD-20-0856.
Weinheimer CJ, Kovacs A, Evans S, Matkovich SJ, Barger PM, Mann DL. Load-dependent changes in left ventricular structure and function in a pathophysiologically relevant murine model of reversible heart failure. Circ Heart Fail. 2018;11(5): e004351. https://doi.org/10.1161/CIRCHEARTFAILURE.117.004351.
Aghajanian H, Kimura T, Rurik JG, Hancock AS, Leibowitz MS, Li L, et al. Targeting cardiac fibrosis with engineered T cells. Nature. 2019;573(7774):430–3. https://doi.org/10.1038/s41586-019-1546-z.
Riehle C, Bauersachs J. Small animal models of heart failure. Cardiovasc Res. 2019;115(13):1838–49. https://doi.org/10.1093/cvr/cvz161.
Cahill TJ, Sun X, Ravaud C, Villa Del Campo C, Klaourakis K, Lupu IE, et al. Tissue-resident macrophages regulate lymphatic vessel growth and patterning in the developing heart. Development. 2021;148(3). https://doi.org/10.1242/dev.194563.
Forte E, Skelly DA, Chen M, Daigle S, Morelli KA, Hon O, et al. Dynamic interstitial cell response during myocardial infarction predicts resilience to rupture in genetically diverse mice. Cell Rep. 2020;30(9):3149–63 e6. https://doi.org/10.1016/j.celrep.2020.02.008.
DeBerge M, Lantz C, Dehn S, Sullivan DP, van der Laan AM, Niessen HWM, et al. Hypoxia-inducible factors individually facilitate inflammatory myeloid metabolism and inefficient cardiac repair. J Exp Med. 2021;218(9). https://doi.org/10.1084/jem.20200667.
Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol A Biol Sci Med Sci. 2014;69(Suppl 1):S4-9. https://doi.org/10.1093/gerona/glu057.
Molawi K, Wolf Y, Kandalla PK, Favret J, Hagemeyer N, Frenzel K, et al. Progressive replacement of embryo-derived cardiac macrophages with age. J Exp Med. 2014;211(11):2151–8. https://doi.org/10.1084/jem.20140639.
Chiao YA, Dai Q, Zhang J, Lin J, Lopez EF, Ahuja SS, et al. Multi-analyte profiling reveals matrix metalloproteinase-9 and monocyte chemotactic protein-1 as plasma biomarkers of cardiac aging. Circ Cardiovasc Genet. 2011;4(4):455–62. https://doi.org/10.1161/CIRCGENETICS.111.959981.
Bruunsgaard H, Skinhoj P, Pedersen AN, Schroll M, Pedersen BK. Ageing, tumour necrosis factor-alpha (TNF-alpha) and atherosclerosis. Clin Exp Immunol. 2000;121(2):255–60. https://doi.org/10.1046/j.1365-2249.2000.01281.x.
Pardali E, Dimmeler S, Zeiher AM, Rieger MA. Clonal hematopoiesis, aging, and cardiovascular diseases. Exp Hematol. 2020;83:95–104. https://doi.org/10.1016/j.exphem.2019.12.006.
Wang Y, Sano S, Yura Y, Ke Z, Sano M, Oshima K, et al. Tet2-mediated clonal hematopoiesis in nonconditioned mice accelerates age-associated cardiac dysfunction. JCI Insight. 2020;5(6). https://doi.org/10.1172/jci.insight.135204.
Khare V, Sodhi A, Singh SM. Effect of aging on the tumoricidal functions of murine peritoneal macrophages. Nat Immun. 1996;15(6):285–94.
Mahbub S, Deburghgraeve CR, Kovacs EJ. Advanced age impairs macrophage polarization. J Interferon Cytokine Res. 2012;32(1):18–26. https://doi.org/10.1089/jir.2011.0058.
Renshaw M, Rockwell J, Engleman C, Gewirtz A, Katz J, Sambhara S. Cutting edge: impaired Toll-like receptor expression and function in aging. J Immunol. 2002;169(9):4697–701. https://doi.org/10.4049/jimmunol.169.9.4697.
Heo GS, Kopecky B, Sultan D, Ou M, Feng G, Bajpai G, et al. Molecular imaging visualizes recruitment of inflammatory monocytes and macrophages to the injured heart. Circ Res. 2019;124(6):881–90. https://doi.org/10.1161/CIRCRESAHA.118.314030.
Brody SL, Gunsten SP, Luehmann HP, Sultan DH, Hoelscher M, Heo GS, et al. Chemokine receptor 2-targeted molecular imaging in pulmonary fibrosis. A clinical trial. Am J Respir Crit Care Med. 2021;203(1):78–89. https://doi.org/10.1164/rccm.202004-1132OC.
Diekmann J, Koenig T, Thackeray JT, Derlin T, Czerner C, Neuser J, et al. Cardiac fibroblast activation in patients early after acute myocardial infarction: integration with magnetic resonance tissue characterization and subsequent functional outcome. J Nucl Med. 2022. https://doi.org/10.2967/jnumed.121.263555.
Hess A, Thackeray JT, Wollert KC, Bengel FM. Radionuclide image-guided repair of the heart. JACC Cardiovasc Imaging. 2020;13(11):2415–29. https://doi.org/10.1016/j.jcmg.2019.11.007.
Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. 2016;375(18):1749–55. https://doi.org/10.1056/NEJMoa1609214.
Mahmood SS, Fradley MG, Cohen JV, Nohria A, Reynolds KL, Heinzerling LM, et al. Myocarditis in patients treated with immune checkpoint inhibitors. J Am Coll Cardiol. 2018;71(16):1755–64. https://doi.org/10.1016/j.jacc.2018.02.037.
Li DK, Wang W. Characteristics and clinical trial results of agonistic anti-CD40 antibodies in the treatment of malignancies. Oncol Lett. 2020;20(5):176. https://doi.org/10.3892/ol.2020.12037.
Ma HS, Poudel B, Torres ER, Sidhom JW, Robinson TM, Christmas B, et al. A CD40 agonist and PD-1 antagonist antibody reprogram the microenvironment of nonimmunogenic tumors to allow T-cell-mediated anticancer activity. Cancer Immunol Res. 2019;7(3):428–42. https://doi.org/10.1158/2326-6066.CIR-18-0061.
Wierz M, Pierson S, Guyonnet L, Viry E, Lequeux A, Oudin A, et al. Dual PD1/LAG3 immune checkpoint blockade limits tumor development in a murine model of chronic lymphocytic leukemia. Blood. 2018;131(14):1617–21. https://doi.org/10.1182/blood-2017-06-792267.
Mazzarella L, Duso BA, Trapani D, Belli C, D’Amico P, Ferraro E, et al. The evolving landscape of “next-generation” immune checkpoint inhibitors: a review. Eur J Cancer. 2019;117:14–31. https://doi.org/10.1016/j.ejca.2019.04.035.
Shrimali RK, Ahmad S, Verma V, Zeng P, Ananth S, Gaur P, et al. Concurrent PD-1 blockade negates the effects of OX40 agonist antibody in combination immunotherapy through inducing T-cell apoptosis. Cancer Immunol Res. 2017;5(9):755–66. https://doi.org/10.1158/2326-6066.CIR-17-0292.
Funding
JJ is supported by the funding provided by R25 HL105400. KJL is supported by the funding provided by the Children’s Discovery Institute of Washington University and St. Louis Children’s Hospital, the Foundation of Barnes-Jewish Hospital, the Burroughs Foundation Welcome Fund, the Leducq Foundation, and the National Institutes of Health (R35 HL161185, RO1 HL151078, R21 AI148877).
Author information
Authors and Affiliations
Contributions
JJ and KJL conceptualized, wrote, and edited the manuscript and figures.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Human and Animal Rights and Informed Consent
All reported studies/experiments with human or animal subjects performed by the authors have been previously published and complied with all applicable ethical standards (including the Helsinki declaration and its amendments, institutional/national research committee standards, and international/national/institutional guidelines).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Myocardial Disease
Rights and permissions
About this article

Cite this article
Jimenez, J., Lavine, K.J. The Dynamic Role of Cardiac Macrophages in Aging and Disease. Curr Cardiol Rep 24, 925–933 (2022). https://doi.org/10.1007/s11886-022-01714-4
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11886-022-01714-4