
Abstract
Heart failure is a consequence of various cardiovascular diseases and associated with poor prognosis. Despite progress in the treatment of heart failure in the past decades, prevalence and hospitalisation rates are still increasing. Heart failure is typically associated with cardiac remodelling. Here, inflammation and fibrosis are thought to play crucial roles. During cardiac inflammation, immune cells invade the cardiac tissue and modulate tissue-damaging responses. Cardiac fibrosis, however, is characterised by an increased amount and a disrupted composition of extracellular matrix proteins. As evidence exists that cardiac inflammation and fibrosis are potentially reversible in experimental and clinical set ups, they are interesting targets for innovative heart failure treatments. In this context, animal models are important as they mimic clinical conditions of heart failure patients. The advantages of mice in this respect are short generation times and genetic modifications. As numerous murine models of heart failure exist, the selection of a proper disease model for a distinct research question is demanding. To facilitate this selection, this review aims to provide an overview about the current understanding of the pathogenesis of cardiac inflammation and fibrosis in six frequently used murine models of heart failure. Hence, it compares the models of myocardial infarction with or without reperfusion, transverse aortic constriction, chronic subjection to angiotensin II or deoxycorticosterone acetate, and coxsackievirus B3-induced viral myocarditis in this context. It furthermore provides information about the clinical relevance and the limitations of each model, and, if applicable, about the recent advancements in their methodological proceedings.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Cardiac inflammation and fibrosis in heart failure
Heart failure (HF) is a chronic disease that is associated with adverse outcome [290]. Due to demographic changes and the increasing prevalence of chronic HF, hospitalisation rates for acute HF are rising [54, 290]. Thus, HF is the leading cause for non-elective hospitalisations in developed countries of patients older than 65 [54]. HF results from injury to the myocardium due to a variety of causes, including, but not limited to, myocardial infarction (MI), hypertension, heart valve disease, and myocarditis [126]. On the one hand, a myocardial injury, regardless of its initiating cause, results in sterile myocardial inflammation [133], on the other hand, the infection with pathogens induces non-sterile myocardial inflammation [284]. In all pathologies, the termination of myocardial inflammation requires the activation of anti-inflammatory stimuli, which in turn initiate pro-fibrotic signalling [60]. Hence, most cardiac pathologies imply the development of myocardial fibrosis [21].
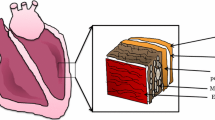
In the healthy heart, the extracellular matrix (ECM) provides a scaffold for cardiac cells and thereby ensures the structural integrity and function of the heart [52]. As shown in Fig. 1a, a decellularised murine heart maintains its morphology, which highlights the overwhelming significance of the ECM for the cardiac integrity. The magnification clearly demonstrates that each cardiomyocyte (CM) is surrounded by ECM, which ensures the transmission of the contractile force from a single CM to the whole organ [133]. Moreover, the ECM provides a reservoir for a variety of latent growth factors [133]. On a pathophysiological level, fibrotic remodelling of the myocardium could disrupt appropriate myocardial excitation–contraction coupling and may promote arrhythmia, e.g. through the induction of re-entry circuits [280]. Furthermore, excessive fibrotic remodelling results in enhanced stiffness and is linked to systolic and diastolic dysfunction [116]. In contrast, dampened fibrotic remodelling in the context of MI may lead to insufficient scar formation, which can be associated with ventricular dilation or even rupture [211, 272].

The cardiac extracellular matrix and its fibrotic remodelling upon injury. a Decellularised murine heart without atria that solely consists of extracellular matrix (ECM) from a macroscopic (left panel) and microscopic (right panel) perspective. The ECM covers every single cardiomyocyte and thus provides a scaffold to conduct the contractile force from each cardiomyocyte to the whole organ and maintains cardiac integrity. b Cross sections of left ventricular tissue (Masson-Trichrome stained) as representative examples of distinct categories of fibrotic extracellular remodelling. Left panel: reactive insterstitial fibrosis, mainly observed in the murine models of nonischemic HF such as transverse aortic constriction, subjection to angiotensin II or deoxycorticosterone acetate, or inoculation with CVB3. Interstitial cardiac fibrosis predominantly occurs as a result of repressing diffuse inflammation upon diffuse cardiac injury. Right panel: reparative fibrosis, mainly observed in the murine models of ischemic HF, such as permanent or transient LAD-ligation. The mechanistic rational of reparative fibrosis is to replace dead cardiac tissue by fibrotic scar tissue. Middle panel: healthy cardiac tissue. Blue: extracellular matrix, pink: cytoplasm
Since pathological fibrosis is characterised by excessive deposition of ECM proteins, several key pro-fibrotic stimuli, such as a variety of growth factors, most importantly transforming growth factor (TGF)-β, but also angiotensin II (Ang II) and aldosterone contribute to the development of cardiac fibrosis regardless of the underlying pathology [133]. However, the relative significance of a distinct molecular pathway depends on the type and the degree of the initial cardiac injury [133]. As shown in Fig. 1b, fibrotic remodelling may be divided into two paramount categories [213]: (1) ‘reactive interstitial fibrosis’, often seen in HF caused by nonischemic injuries and (2) ‘reparative fibrosis’, often seen in HF caused by prolonged ischemia to replace necrotic CMs by the formation of a myocardial scar.
Several murine models of HF exist, which either predominantly develop reparative or reactive interstitial fibrosis. Sterile inflammation and reparative fibrosis upon ischemia are observed in the murine models of (1) permanent or (2) transient ligation of the left anterior descending coronary artery (LAD). Sterile inflammation and reactive interstitial fibrosis upon pressure overload and neurohumoral activation are observed in the murine models of (1) transverse aortic constriction (TAC), (2) chronic infusion of angiotensin II (Ang II), and (3) chronic treatment with deoxycorticosterone acetate (DOCA) accompanied by unilateral nephrectomy and high salt diet. Non-sterile, excessive inflammation and subsequent interstitial fibrosis occurs in the murine model of viral myocarditis upon infection with coxsackievirus B3 (CVB3). Depending on the susceptibility of a certain mouse strain, viral myocarditis may further become chronic and thus result in dilated inflammatory cardiomyopathy.
This review aims to facilitate the challenging selection of a proper murine model of HF for a specific research question as it (1) provides an overview about the distinct molecular and cellular mechanisms that underlie cardiac inflammation and fibrosis in each of these six frequently used HF models. Further, it aims to provide information about (2) the methodological proceeding, (3) the clinical relevance, (4) the limitations and, if applicable, (5) the recent methodological advancements of each model.
Models of heart failure induced by ischemia
Myocardial infarction induced by permanent LAD-ligation
Murine model of heart failure and its clinical relevance
Permanent LAD-ligation in mice results in acute myocardial injury due to ischemia, which is defined as MI [163, 256, 289]. In humans, type 1 MI is caused by the disruption of an atherosclerotic plaque and results in myocyte necrosis, which is often accompanied by ST-elevations in the ECG (ST-elevated MI, STEMI). According to the ESC guidelines, prompt revascularisation for STEMI patients is recommended to reduce infarct size and mortality, but still a significant number of STEMI patients (approximately 15–25%) do not receive any form of reperfusion therapy [33, 76]. The current ESC guidelines for the treatment of STEMI additionally state that reperfusion is not recommended if hemodynamically stable patients present more than 12 h after the onset of symptoms [33, 110]. Thus, permanent LAD-ligation in mice particularly resembles the clinical situation of a significant fraction of patients that suffer from type 1 MI without timely revascularisation [256]. Even patients that undergo revascularisation can develop a form of ischemic cardiomyopathy resembling permanent LAD-ligation in animal models. In mice, the development of HF following permanent LAD-ligation is characterised by infarct wall thinning, left ventricular dilation and severe systolic and diastolic dysfunction. These hallmarks are frequently seen in post-MI patients as well. Since MI in mice is predominantly characterised by ventricular dilation, their HF resembles features of human HF with reduced ejection fraction (HFrEF) [94, 163, 273]. HFrEF in mice is observed within the first 24 h after MI and further aggravates over time [114, 187].
Methodological proceedings
Induction of HF utilising the model of permanent LAD-ligation in small rodents was first described in 1979 in rats [207]. Initially, adult mice are anaesthetised and mechanically ventilated. Hereafter, surgical access to the heart is performed by an incision through the third left intercostal space [94]. Subsequently, the LAD is permanently ligated with a suture [94]. Figure 2a shows a schematic drawing and an intraoperative image of the procedure and the sufficient ligation, proven by blanching of the distal cardiac tissue [130]. Moreover, infarct-typical ECG-alterations, such as ST-elevation [75, 86], or elevated plasma levels of cardiac troponin in the cause of MI [1] can prove the ischemia-induced MI. The observation period following permanent LAD-ligation in mice typically ranges from a few hours up to several weeks [286].

Myocardial infarction induced by ligation of the left anterior descending artery (LAD) in mice. a Left: schematic drawing that depicts the ligation of the LAD in mice and the resulting area at risk (white). Right: representative intraoperative image immediately taken after the LAD-ligation, in which the area at risk is demasked by the blanching of the tissue distal to the ligation. The size of the area at risk depends on the area of cardiac tissue that is supplied by the LAD. b Time course of the wave front of CM-necrosis (infarct zone, dark grey) within the area at risk (white) in mice. Ischemia of more than 20–30 min results in CM-necrosis, which is initially observed in the subendocardial layers and hereafter extends to the subepicardial layers; ultimately, after more than 60–90 min of ischemia, this results in the total infarction of the area at risk
To uncover the infarct zone of explanted hearts, the cardiac tissue can be incubated with the colourless, hydrophilic triphenyltetrazolium chloride immediately after harvesting. In viable cells, this dye is reduced to a deep red, hydrophobical formazan. Thus, only areas that contain viable CMs appear in deep red, whereas the infarct zone itself remains colourless [94, 155]. The mortality following permanent LAD-ligation in mice predominantly occurs from day 1 to day 7 as a result of left ventricular rupture, acute HF, or heart rhythm disturbances [39, 70, 86, 130, 156, 187].
Myocardial infarction induced by transient LAD-ligation (ischemia–reperfusion)
Murine model of heart failure and its clinical relevance
The murine model of transient LAD-ligation mimics type 1 MI followed by successful revascularisation [155], which reflects the clinical course of the majority of STEMI patients. According to the ESC guidelines, primary PCI, defined as percutaneous catheter intervention (PCI) without previous fibrinolysis, is recommended as the reperfusion strategy for STEMI patients within 90 min after diagnosis [110, 195]. Several clinical trials emphasise the strong association between delayed revascularisation and the degree of myocardial injury and its sequelae [109, 184, 256]. Mice that are subjected to transient LAD-ligation exhibit HF symptoms comparable to permanent LAD-ligation, such as dilated chamber dimensions, and thus resemble features of human HFrEF [257]. However, the severity of HF induced by transient LAD-ligation highly depends on the time of ischemia. As depicted in Table 1, the degree of cardiac dysfunction and the size of the infarct zone are typically less pronounced in ischemia–reperfusion (I/R) models than in models of permanent LAD-ligation [179]. Next to the ischemic damages in the I/R model, subsequent reperfusion exposes the cardiac tissue to oxidative stress, which exaggerates the tissue damage; this additional injury is entitled reperfusion injury [184, 256]. Thus, murine I/R models are likewise used for investigations that focus on the pathophysiology of this reperfusion injury.
Methodological proceedings
The surgical preparation for the transient LAD-ligation in mice is performed similar to the procedure of inducing permanent LAD-ligations. Commonly, one-step procedures are performed in which the surgical preparation is immediately followed by the transient LAD-occlusion [179]. A sufficient occlusion may be verified by blanching of the cardiac tissue distal to the ligation, as shown in Fig. 2a, or by an ST-elevation in the ECG [120]. In the literature, ligation is maintained for 20–60 min [120, 178, 202, 257, 287]. However, LAD-ligation of less than 30 min may not provoke ischemia-induced myocardial injury, whereas ligation of 60–90 min results in irreversible CM death and complete infarction of the area at risk as depicted in Fig. 2b [155]. Thus, 45–60 min of ischemia is recommended for the transient LAD-ligation. The observation period after subsequent reperfusion ranges from a few minutes up to several weeks [120, 178, 202, 257, 287], depending on the respective scope of the study.
The area at risk is defined as the area of non-perfused cardiac tissue, in which the wave front of the infarct zone subsequently extends (Fig. 2b). The size of the area at risk following transient LAD-ligation can be estimated by injecting Evans Blue into the circulation. This dye is subsequently distributed to the perfused tissue but leaves the non-perfused area at risk unstained. Perfusing the heart with Evans blue can either be performed in vivo or ex vivo [155, 273]. Afterwards, the size of the infarct zone can be estimated similar to permanent LAD-ligation using the triphenyltetrazolium chloride staining in the harvested cardiac tissue.
Recent advancements
LAD-ligation protocols are surgical procedures that, by themselves, may induce systemic inflammatory responses. To avoid that these systemic inflammatory responses interfere with local inflammatory processes during cardiac repair, the procedure may be split into a preparative surgery which is followed by the transient LAD-ligation several days after the initial surgery [120, 197]. Furthermore, the use of volatile anaesthetics during ischemia can precondition the murine heart to face reperfusion injury for up to 72 h [213]. Therefore, splitting the procedure avoids this preconditioning. However, systemic inflammatory responses after LAD-ligations may be limited to rather invasive procedures that include, e.g. thoracotomies and may not be seen in minimally invasive procedures. In line with this, a recent study concludes that minimally invasive sham LAD-ligations induce several pro-inflammatory genes but none of these genes passes the threshold required for increased protein expression [114].
Distinct characteristics of post-infarct remodelling in models of permanent and transient LAD-ligation
The degree and the temporal dynamics of post-infarct remodelling in mice depend on a proximal or distal ligation of the LAD and the use of reperfusion protocols [285]. Compared to permanent LAD-ligation, ischemia/reperfusion (I/R) subjects murine hearts to (1) a limited ischemic period and (2) a restored blood flow which allows effector cells to quickly take hold of the affected area and provides metabolites to the cells in the infarct wall. As a consequence, murine I/R models possess specific features that make them different from models of permanent LAD-ligation. These differences are summarised in Table 1. In general, murine I/R induces smaller infarct areas that are primarily located midmyocardial as compared to the transmural infarct areas observed after permanent LAD-ligation [93, 119, 285]. Further, reperfusion lowers the degree but accelerates the temporal dynamics of the postischemic remodelling [4, 285]. This is associated with less fibrotic remodelling and less impaired contractile function in the chronic phase after MI in I/R models [4, 285]. Beyond that, the time point of reperfusion may crucially affect the relative contributions of distinct cellular effectors to the postischemic pool of activated interstitial cells [4]. Due to their loss in the infarct zone, prolonged ischemia favours the recruitment of interstitial cells from the non-infarcted tissue or the blood stream, whereas quick reperfusion may favour the activation of viable resident myocardial cells [4].
Cardiac inflammation and fibrosis induced by ischemia
In mammals, adult CMs possess negligible self-regenerative capacity [53, 186]. Hence, cardiac remodelling after MI ultimately results in the formation of a fibrotic scar. This process of reparative fibrosis consists of three sequential stages [190, 213, 245]: (1) during the initial inflammatory phase (acute phase), necrotic CMs release danger signals; this rapidly activates resident sentinel cells, which initiate the recruitment of pro-inflammatory cells to clear the infarct wall from the cellular debris and the defective ECM [61, 190]. The sufficient clearance of necrotic CMs, ECM fragments and apoptotic immune cells in the infarct wall promotes the resolution of inflammatory signals and thus the transition to the proliferative phase. (2) During the subsequent proliferative phase (subacute phase), cardiac fibroblasts (CFs) transdifferentiate into myofibroblasts (MFs) and secrete an abundant amount of collagen and non-structural matrix proteins to compensate the cellular loss [133, 190]. (3) During the maturation phase (chronic phase), the immature fibrillar collagens that have been secreted during the preceding proliferative phase are enzymatically cross-linked and thereby gain stability, whereas the cellular effectors mostly vanish [133, 190]. The degree of inflammation and fibrosis following MI in mice varies throughout distinct regions in the cardiac muscle, depending on their proximity to the infarct zone. Therefore, in models of murine MI the left ventricle is most often divided into three distinct zones: infarct zone, border zone and remote zone. In general, inflammation and fibrosis are most pronounced in the infarct zone, followed by the border zone, but can also spread into the remote zone in severe cases [94]. Table 1 provides an overview of the distinct temporal dynamics of cardiac inflammation and fibrosis in the murine models of permanent and transient LAD-ligation.
Acute phase after MI—the inflammatory phase
The inflammatory phase after MI plays an ambiguous role, as it is a pivotal basis of cardiac repair but also a crucial factor for a subsequent development of HF. Thus, the local and temporal restriction of inflammation is crucial for sufficient infarct healing. A schematic drawing of the tightly regulated interplay between pivotal cellular and molecular players of the inflammatory phase and its subsequent resolution is shown in Fig. 3.

The initiation of inflammation and its subsequent resolution in the context of myocardial infarction. Ischemia induces necrosis of cardiomyocytes (nCM) and thus the release of danger signals, such as mitochondrial (mt)DNA, the chromatin protein HMGB1, purine metabolites, sarcomeric fragments, S100 proteins and reactive oxygen species (ROS). Subsequently, resident sentinel cells, such as endothelial cells (ECs), cardiac fibroblasts (CFs) and macrophages (MPs) are activated (a) by their pattern recognition receptors (PRRs) and secrete pro-inflammatory mediators, e.g. interleukins (ILs), chemokines (CCLs), and tumor necrosis factor (TNF)-α. Further, ECs express cell-adhesion molecules (CAMs) upon activation and thereby facilitate leukocyte recruitment. Hereafter, leukocytes invade the injured tissue and are immediately confronted with the pro-inflammatory environment. Hence, they gain a pro-inflammatory signature (M1-polarised macrophages and N1-polarised neutrophils) and further amplify the inflammatory response by secreting pro-inflammatory cytokines, but also matrix metalloproteinases (MMPs). MMPs cleave the matrix and thus generate matrix fragments (matrikines) which additionally serve as pro-inflammatory stimuli. Subsequently, M1-MPs and aCFs clear the wound from necrotic CMs but also from apoptotic neutrophils and matrikines and thereby resolve the pro-inflammatory environment. As they passage to the proliferative phase, the effector cells of the inflammatory phase undergo a second reprogramming and gain a reparative signature, by which they become myofibroblasts (MFs), N2-polarised neutrophils and M2-polarised MPs. Subsequently, these phagocytes, together with regulatory T-cells (Tregs), release anti-inflammatory stimuli such as transforming growth factor (TGF)-β1, IL-10, lactoferrin, annexin A1, lipoxins and resolvins which further repress inflammation and provide a proliferative milieu. During the subsequent proliferative phase, predominantly MFs, but also M2-polarised MPs secrete abundant ECM proteins, e.g. collagens and matricellular proteins and thereby promote the formation of the myocardial scar
Initiation of inflammatory cascades—danger signals, sentinel cells and pro-inflammatory mediators
MI is pathologically defined as myocardial cell death due to prolonged ischemia [256]. Necrotic CMs release danger signals, the so-called damage-associated molecular patterns (DAMPs), such as mitochondrial DNA (mtDNA), the chromatin protein high mobility group box 1 (HMGB1), purine metabolites, sarcomeric protein fragments and S100 proteins [19, 57, 136, 158]. However, the cellular specificity of DAMPs is poorly understood [57]. They initially alarm a variety of sentinel cells, such as resident macrophages (MPs), endothelial cells (ECs), resident cardiac fibroblasts (CFs), surviving CMs in the border zone and subsequently also invading leukocytes [57]. The main pattern recognition receptors (PRRs) of the cardiac sentinel cells are (1) a variety of toll-like receptors (TLRs), (2) the receptor of advanced glycation end-products (RAGE) and (3) NOD-like receptors (NLRs) [211]. PRRs are either localised in intracellular compartments or on cellular surfaces. Therefore, specific PRRs play distinct roles in phagocytes and non-phagocytes. Recently, interactions of DAMPs and their corresponding PRRs in the context of MI have been comprehensively reviewed [211]. Downstream PRR-signalling activates the transcription factor nuclear factor kappa B (NFκB), which subsequently elevates the expression of chemokines, cytokines and cell-adhesion molecules (CAMs) in the infarct wall [61, 80, 168, 211]. While secreted chemokines attract leukocytes, CAMs exposed on ECs facilitate their invasion [211]. Distinct chemokines determine the recruitment and composition of specific immune cells in the myocardium: while CC chemokines, such as CC-chemokine ligand (CCL)-2, -5, and -7, attract monocytes, lymphocytes and mast cells, CXC chemokines, such as CXCL-8, attract neutrophils [61, 133]. Pro-inflammatory cytokines, such as TNF-α and the interleukins (IL)-1β, -6, and -18, further amplify the inflammatory response by activating resident and invading effector cells [211]. In this context, interleukin (IL)-1β may play a crucial role as it mediates myocardial chemokine synthesis and induces a pro-inflammatory M1 polarisation in MPs [22, 61, 133]. In line with this, IL-1 receptor type I deficient mice exhibit less inflammation and less fibrosis following MI [22]. Both, leukocytes and activated CFs, secrete the mature form of IL-1β; this requires the preceding caspase-1-mediated cleavage of its precursor pro-IL-1β in a multiprotein complex entitled ‘inflammasome’; the complex activation of inflammosomes is comprehensively reviewed elsewhere [233]. In the early phase after MI, IL-6 upregulates the expression of hyaluronan-synthases, which in turn provides a basis for MF transdifferentiation and thus cardiac repair; hence, IL-6 blockade may be detrimental after MI [188].
Oxidative stress and the reperfusion injury
Next to the irreversible CM-necrosis in the infarct zone, the transient depletion of oxygen and other metabolites in the area at risk switches the metabolism of viable cells to anaerobic glycolysis and disrupts appropriate ion homeostasis [88]. This results in intracellular acidosis and accumulation of Ca2+ and Na+ [88]. During acute ischemia, high intracellular Na+ and H+ protect the cell from mitochondrial damage as they close the mitochondrial permeability transition pore (MPTP) and prevent the deteriorating effects of myofibril hypercontracture [88]. Upon reperfusion, oxidative phosphorylation and acid–base balance are rapidly restored within cells of the area at risk [88]. This leads to (1) opening of the mitochondrial permeability transition pore (MPTP) and thus mitochondrial damage, (2) myofibril hypercontracture and (3) excessive accumulation of reactive oxygen species (ROS) [88]. ROS further aggravate the reperfusion injury as they induce the dysfunction of the sarcoplasmic reticulum, oxidise membrane lipids and DNA and promote endothelial dysfunction [88, 170]. Moreover, ROS potently attract leukocytes [88, 170]. Accordingly, reperfusion of the area at risk following ischemia induces an additional reperfusion injury, which is of particular importance in the murine model of transient LAD-ligation. The current understanding of the pathophysiology of the reperfusion injury is reviewed in more detail elsewhere [88, 92].
Initial matrix remodelling and the first provisional matrix
Activated resident cells, especially CFs and MPs, as well as recruited leukocytes secrete and activate matrix metalloproteinases (MMPs) of both subgroups, the collagenases (e.g. MMP-8 and -13) and the gelatinases (e.g. MMP-2 and -9) [58]. Recently, the role of MMPs following MI has been comprehensively reviewed [154]. By degrading ECM proteins, MMPs generate matrix fragments (matrikines) which additionally serve as pro-inflammatory stimuli [133]. Well-characterised matrikines are (1) proline–glycine–proline (PGP), a collagen fragment which attracts neutrophils through binding to their CXC-receptor-1 and -2 and (2) a fragment of the glycosaminoglycane hyaluronan, whose delayed clearance is associated with excessive inflammation [58, 69, 264]. Beyond pro-inflammatory features, distinct matrikines may also orchestrate proliferative cellular responses, as, e.g. a fragment of collagen XVIII (endostatin) enhances fibroblast proliferation [58]. A plasma-derived, provisional matrix initially replaces the fragmented ECM; rapid upregulation of vascular endothelial growth factor (VEGF) in the infarct wall increases vascular permeability, which leads to the extravasation of the plasma proteins fibrin and fibronectin [5, 58]. This fibrin-based provisional matrix initially stabilises the infarct wall and provides a scaffold for leukocyte invasion [35, 44, 48]. Furthermore, through interactions with cellular integrins, it may alter the inflammatory profile of migrating leukocytes and CFs [58]. However, the specific regulatory effects of the early fibrin-based matrix remain poorly understood.
Endothelial cells
Despite the common understanding of the cellular composition of the non-myocyte fraction in the mammalian heart during the last decades, recent findings revealed that ECs are the most abundant non-myocytes (> 60%) in the healthy murine heart [209]. This changed the preceding doctrine that CFs are the most abundant ones. Upon stimulation with DAMPs and pro-inflammatory cytokines, ECs facilitate leukocyte extravasation into cardiac tissue [211]. Therefore, they (1) immediately mobilise preformed P-selectin from their weibel-palade bodies [211] and (2) subsequently upregulate the expression of CAMs, such as ICAM, VCAM and selectin on their cell surface. Further, ECs are a relevant source of cytokines and chemokines after MI [138, 292]. Recently published work shows that the pro-inflammatory activation of ECs after MI may be triggered by the CM-dependent release of natriuretic peptides, such as ANP and BNP [28]. This release is an immediate response after MI and correlates with the severity of the event [28]. Since ECs immobilise chemokines to glycosaminoglycanes on their surface, ECs also play a pivotal role for the local concentration of leukocyte attractors in the injured myocardium [61, 212].
Cardiac fibroblasts
In the healthy murine heart, resident CFs represent less than 20% of non-myocytes [209]. Nonetheless, after MI their population undergoes marked expansion. In spite of the debate surrounding the origin of activated CFs in the infarcted murine heart, there is a growing body of evidence that the majority of activated CFs during MI originates from resident interstitial Tcf21+ fibroblasts [4]. In vitro experiments showed that CFs secrete pro-inflammatory mediators (IL-1β, -1α, -6, -8, and TNF-α) and matrix-degrading proteases (MMP-2, -3, -9, -12) upon stimulation with DAMPs or ROS [59, 154, 243]. However, their in vivo role in initiating cardiac inflammation after MI has just recently been shown. In isolated CFs, the loss of the G protein-coupled receptor kinase (GRK)-2 attenuates the expression of pro-inflammatory cytokines [278]. Thus, recent investigations in mice with a fibroblast-specific depletion of the G protein-coupled receptor kinase (GRK)-2 provide the first direct in vivo evidence for the pro-inflammatory role of CFs in this context, as these mice exhibit less neutrophils in their infarct walls, smaller infarct sizes and better contractile function 24 h after I/R [278]. The physiological signification of the pro-inflammatory feature of CFs may be that the secretion of inflammatory cytokines, especially IL-1β, prohibits premature MF transdifferentiation [229] and might therefore ensure the proper clearance of the infarct wound before scarring [59]. Beyond that, CFs mediate CM-apoptosis by secreting (1) pro-apoptotic inflammatory cytokines, (2) matrix-degrading MMPs, which deprive a sufficient ECM-environment, and (3) pro-apoptotic exosomes [59].
Neutrophils
Neutrophils are the first cellular responders [61] and invade especially the border zone, mainly attracted by CXC chemokines [57, 211]. In comparison to other leukocytes, their number increases immediately after MI. While the highest neutrophil/leukocyte ratio (25%) in the infarct zone is seen at day 1, their total number increases until day 3 after MI (Table 1) [285]. Subsequently, the neutrophil/leukocyte ratio decreases to less than 5% at day 5 after MI [285]. Recent findings reveal that the degree of neutrophil recruitment into the infarcted cardiac tissue in mice shows a circadian kinetic, as it depends on the time-of-day onset of MI [232]. Confronted with the pro-inflammatory environment, infiltrating neutrophils exhibit an inflammatory N1-phenotype, which depends on the DAMP-mediated activation of TLR-4 [167]. Neutrophils play ambiguous roles during the post-MI remodelling: On the one hand, the number of inflammatory neutrophils positively correlates with infarct wall thinning in mice, which was related to their secretion of MMP-12 and -25 [167]. Furthermore, neutrophils secrete highly toxic mediators, such as myeloperoxidase, which worsen the postischemic remodelling and promote arrhythmia [182]. Beyond their direct pro-inflammatory actions, neutrophils mediate the recruitment of monocytes [57]. Recent findings reveal that this happens in a synergistic cooperation with platelets, as human neutrophil peptide (HNP)-1 needs to form heteromers with the platelet-derived chemokine CCL-5 to fulfil its functions [3]. On the other hand, the recruitment of early inflammatory N1-neutrophils is essential for their subsequent N2-polarisation [100]. N2-polarised neutrophils were recently identified to contribute to the reparative M2-polarisation of MPs during the proliferative phase [100] and may thus be essential for a sufficient formation of the scar.
Monocytes/macrophages
A relatively low number of non-myocytes in the healthy cardiac tissue are assigned as leukocytes (approx. 7%); however, the vast majority of these cells are identified as resident MPs [49, 209, 291]. Thus, resident MPs play a crucial role as sentinel cells that initiate cardiac inflammation early after MI. Moreover, their absolute numbers increase following MI [285]. However, as neutrophils exhibit faster kinetics in invading the cardiac tissue during the acute phase, the MP/leukocyte ratio declines to approx. 60% until day 3 after MI [285]. Hereafter, further monocytes invade, whereas neutrophils undergo apoptosis; thus the MP/leukocyte ratio is reconstituted to more than 80% between day 7 and 14 after MI [285]. Hence, the cardiac recruitment of tissue-invading pro-inflammatory Ly6Chi-monocytes, which later differentiate into MPs, is a key event in postischemic remodelling. The recruitment of monocytes is mediated in four sequential stages: (1) MI alters the hemodynamic conditions and thereby promotes the release of catecholamines from the adrenal glands into the blood stream. (2) high plasma levels of catecholamines repress stem-cell retention factors (e.g. CXCL-12) in bone marrow and spleen stem cell niches and thereby enable monocytes to leave their niches and enter the blood stream [48]. (3) the upregulated CCL-2 and CCL-7 expression in the injured myocardium attracts these pro-inflammatory monocytes and subsequently (4) mediates their invasion into the cardiac tissue [48]. The mechanistic significance of the initial catecholamine release from the adrenal glands is highlighted, as adrenergic β2-receptors are pivotal for the expression of monocytic CC-chemokine-receptor (CCR)-2 and thus for monocyte recruitment [81, 82].
Hereafter, MP colony-stimulating factor (M-CSF) promotes the differentiation of invading monocytes into MPs [63]. Once they are differentiated, MPs are confronted with the hypoxic environment in the infarct wall. This promotes the expression of hypoxia-inducible factor (HIF)-1, which alters MP metabolism towards glycolysis, a key feature of M1-polarisation [187]. Further, the high concentration of DAMPs, IL-1β and TNF-α in the infarct wall contributes to their pro-inflammatory M1 polarisation [187]. Early M1 MPs secrete and activate MMPs and can thereby dig through the infarct zone to engulf and remove necrotic CMs [133, 154, 187]. However, to limit excessive activation of inflammatory pathways in these early M1 MPs, several anti-inflammatory genes, such as schlafen (Slfn)-4 and CD-9, are concomitantly upregulated [187].
Mast cells
The key feature of mast cells is their high cytoplasmic density of preformed granules. Thus, they are able to immediately respond to external stimuli. Despite that the number of mast cells may not be significantly increased after MI in mice [43], they are an important source of TNF-α and thus initiate the cytokine cascade and may orchestrate cardiac inflammation [62, 127]. However, mast cells play an ambiguous role in the postischemic remodelling: on the one hand, mast cell-deficient mice [145], as well as mice with a knockout of the mouse mast cell protease (mMCP)-4 [101], exhibit improved prognosis after MI. In the context of MI, the secretion of the chymase mMCP-4 promotes cardiac inflammation and subsequent fibrotic remodelling by activating IL-1β, angiotensin II, endothelin-1, MMP-9 and fibronectin [101]. On the other hand, mast cell deficiency has been shown to be beneficial in mice within the first hours after MI [18], but to induce larger infarct sizes and accentuated ventricular dilation during the chronic phase [30]. Further, a recent study concludes that mast cells preserve contractile function during MI by upregulating the Ca2+-sensitivity of myofilaments [196]. Thus, the role of mast cells in cardiac postischemic remodelling is still under debate.
Lymphocytes
For decades, investigations focused on the role of the innate immunity in the postischemic remodelling. In contrast, the role of the adaptive immunity in postischemic remodelling following MI has been subject to less scrutiny [99]. Recently, the role of the adaptive immunity in the context of MI has been comprehensively reviewed [99]. Briefly, the invasion of adaptive immune cells into cardiac tissue reaches a peak at day 7 [99, 285]. In this context, the depletion of B-cells has been shown to be beneficial in murine models of MI [99]. However, knockout studies revealed that the depletion of CD4+ T-cells deteriorates cardiac function and worsens cardiac remodelling after MI in mice [99]. CD4+ T-cells in postischemic remodelling are predominantly Th1-polarised, whereas Th2- and Th17-polarised T-cells are not increased following MI [99, 285]. The role of regulatory T-cells (Tregs) in postischemic remodelling is highlighted as the depletion of Tregs deteriorates and the stimulation of Tregs vice versa ameliorates adverse cardiac remodelling [99, 253]. In general, Tregs downscale cardiac inflammation (1) by secreting anti-inflammatory mediators (e.g. IL-10 and TGF-β1), (2) by cell-to-cell interactions that inhibit pro-inflammatory T-cells and (3) by cell-to-cell interactions that inhibit antigen presentation by, e.g. dendritic cells [177]. The specific role of Tregs in cardiovascular disease has recently been reviewed comprehensively [177].
Resolution of cardiac inflammation and transition to the proliferative phase
The resolution of cardiac inflammation is mandatory for the transition to the proliferative phase and thus for a sufficient formation of the scar. The resolution of inflammation is associated with the clearance of dead tissue and the subsequent reprogramming of effector cells which in turn release anti-inflammatory molecules.
Removal of inflammatory stimuli and reprogramming of phagocytes
Efferocytosis is the phagocytosis of dead cells and a key signal that dampens and finally resolves the inflammation in the injured myocardium after MI [211]. Efferocytosis implies the removal of (1) necrotic CMs in the infarct zone, (2) apoptotic CMs in the border zone, and (3) apoptotic leukocytes, especially neutrophils, which undergo apoptosis owing to their short life-span of 3–7 days [61, 194, 211]. Sufficient efferocytosis and thus sufficient resolution of cardiac inflammation depends on the myeloid-epithelial-reproductive protein tyrosine kinase (MERTK) and the milk fat globule-EGF factor 8 (MFG-E8) [102]. Moreover, MFs also contribute to the resolution of inflammation as they engulf dead cells similar to MPs in a MFG-E8-dependent manner [193]. The level of MF-dependent efferocytosis is almost 40% of that of MPs [193]. Moreover, MPs internalise matrikines and thereby eliminate further pro-inflammatory stimuli [210]. However, efferocytosis does not only clear the wound from pro-inflammatory stimuli, but also promotes a reprogramming of MPs; initially, the early pro-inflammatory M1 MPs reprogramme and become intermediate MPs, which already perform oxidative phosphorylation, a key feature of M2-polarisation [187, 220]. Thus, the expression of AMP-activated kinase (AMPK), which mediates the metabolic conversion from glycolysis to oxidative phosphorylation [266], may display a crucial factor for MP reprogramming [187]. During further progression from the inflammatory to the proliferative phase, MPs undergo a second reprogramming by which they gain a reparative M2-signature. This second reprogramming depends on an IL-6 and IL-10 initiated pathway, the signal transducer and activator of transcription (STAT)-3 pathway. Additionally, TGF-β and IL-4 initiated signalling is required in this context [187, 263]. Similar to MPs, MFs acquire an anti-inflammatory signature during the progression from the inflammatory to the proliferative phase in an efferocytosis-dependent manner [193].
Key mediators of anti-inflammatory, pro-fibrotic signalling
Several anti-inflammatory and concomitantly pro-fibrotic mediators orchestrate the transition from the inflammatory to the proliferative phase: (1) apoptotic neutrophils release lactoferrin and annexin A1, mediators that inhibit further leukocyte invasion [61]. (2) efferocytosis of dead cells stimulates MPs to secrete anti-inflammatory TGF-β1, IL-10 and anti-inflammatory lipids like lipoxins and resolvins [61, 108, 246]. (3) Tregs additionally secrete TGF-β1 and IL-10 [57]. (4) MMPs limit leukocyte invasion by cleaving CCLs [274], which converts the pro-inflammatory leukocyte attractors to anti-inflammatory chemokine receptor antagonists and thus prohibits further leukocyte invasion [77, 275]. Furthermore, MMPs cleave CCL binding sites on ECs, the glycosaminoglycanes, which prohibits local chemokine accumulation [77, 274]. (5) heparin-binding domains on fibrin molecules of the provisional matrix provide a reservoir for anti-inflammatory, proliferative growth factors, such as TGFs, PDGFs, FGFs, and VEGFs [171].
Subacute phase after MI—the proliferative phase
The proliferative phase after MI is characterised by the proliferation of interstitial cells, a highly dynamic ECM and the vascularisation of the granulation tissue.
TGF-β1 and the transdifferentiation of cardiac fibroblasts
The pivotal mediator of the proliferative phase is thought to be TGF-β1 [56]. Though there are three TGF-β isoforms, TGF-β1 is the major and best characterised isoform in the cardiac tissue [56]. TGF-β1 strongly represses inflammation and matrix degradation but enhances ECM enlargement and cell proliferation. It stimulates the anti-inflammatory polarisation of leukocytes and the transdifferentiation of CFs [41]. Furthermore, it induces anti-inflammatory protease inhibitors, such as plasminogen activator inhibitor (PAI)-1 and tissue inhibitors of MMPs (TIMPs) [133, 231]. In the healthy heart, TGF-β1 is bound to the ECM as an inactive, latent complex [6, 222]. The release of the bioactive form from latent stores is mediated by proteases like plasmin [165, 222] and MMPs, such as MMP-2 and MMP-9 [288]. Moreover, matricellular proteins, ROS and the acidic environment promote the release of mature TGF-β1 from its latent form after MI [12, 165]. Beyond the mobilisation of TGF-β1 from latent stores, CFs, ECs, MPs, and platelets actively secrete mature TGF-β1 [56]. TGF-β1 binds to its type II receptor (TβRII) on the cell surface of various target cells; subsequently the receptor–ligand complex transphorylates the cytoplasmic domain of the type I receptor (TβRI) [242]. Hereafter, downstream intracellular signalling involves Smad2/3-dependent as well as Smad-independent pathways [133, 242]. TGF-β1 requires the concomitant presence of EDA-fibronectin to induce MF transdifferentiation [9, 238] by enhancing the Smad3-dependent α-smooth muscle actin (α-SMA) expression in CFs [45, 68, 97]. Beyond that, Smad3-signalling in MFs crucially regulates the integrin-mediated formation of organised MF arrays [134]. Furthermore, hyaluronan significantly contributes to MF transdifferentiation, as the loss of its main receptor CD44 results in lower collagen secretion after murine MI [106]. Though lineage tracing identified resident CFs as the major source of MFs [4], pericytes, ECs as well as circulating fibrocytes of bone marrow origin may also contribute to the population of MFs in cardiac repair [133, 183]. Key features of MFs are a highly active endoplasmic reticulum and α-SMA-containing stress fibres, which allow their contraction [95, 96]. Their stress fibres are connected to specific domains of extracellular fibronectin in an adhesion complex called ‘fibronexus’ [52]. Thereby, MFs are able to exert tensile force on the surrounding ECM [52]. This may explain the reduction of the scar size as compared to the initial infarct size by about 50% [262]. During the proliferative phase, MFs undergo a rapid expansion primarily in the border zone and secrete structural proteins, such as the fibrillar collagen-I and -III [31, 265]. Additionally, MFs secrete matricellular proteins upon stimulation with several growth factors or angiotensin II [58, 95]. Next to MFs, reparative M2 MPs were recently shown to deposit ECM proteins during the proliferative phase [187].
The second provisional matrix
During the proliferative phase, the secretion of plasminogen and its subsequent activation disintegrate the provisional, fibrin-based matrix [36]. In line with this, plasminogen-deficient mice exhibit an impaired formation of the granulation tissue after MI (61). The first plasma-derived matrix is subsequently replaced by a second, cell-derived one: MFs and MPs mainly secrete fibronectin [20], but also hyaluronan, proteoglycans and matricellular proteins [58]. Since distinct growth factors, most importantly TGF-β1, require interactions with matrix components for their signal transduction, the cell-derived matrix becomes a conductor of the remodelling orchestra during proliferative phase [58]. Further, the requirement for a cooperation between matrix components and growth factors restricts the proliferative signalling to the infarct wall [58].
Matricellular proteins
Matricellular proteins display their highest expression in the border zone and serve as molecular bridges between ECM proteins and surface receptors [58]. Thereby, they limit inflammation and induce reparative reprogramming in interstitial cells [58]. Further, their requirement for growth factor and cytokine signalling prevents excessive fibrotic remodelling after MI since they localise proliferative stimuli to the infarct wall and temporarily restrict pro-fibrotic signals [58]. Well-characterised matricellular proteins are, e.g. thrombospondins (TSPs), tenascins, the CCN-family, periostin, osteopontin, and secreted protein acidic and rich in cysteine (SPARC) [58, 133, 221]. TSP-1 and -2 are markedly upregulated in the border zone after MI [58]. They activate TGF-β1 and inhibit MMP-2 and -9, by which they build a barrier against the diffusion of pro-inflammatory molecules into the remote zone [64, 221]. CCN-2 has TGF-β1 synergistic, proliferative effects on interstitial cells, but also promotes CM hypertrophy [213]. Osteopontin prevents excessive cardiac dilation by stimulating collagen deposition into the infarct zone [258]. The depletion of SPARC in the context of MI, however, results in higher rates of cardiac rupture and cardiac dysfunction due to immature collagen and disorganised granulation tissue [230]. Beyond that, the clearance of matricellular proteins supresses proliferative stimuli and thereby promotes the transition from the proliferative phase to the maturation phase [58]. In this context, the stabilin-1-mediated phagocytosis of SPARC is a well elaborated signal [140].
Vascular remodelling
Vascular remodelling is a major process during the proliferative phase. Sprouting of former quiescent ECs upon hypoxia and high concentrations of VEGF induce the formation of neovessels [32, 61, 146]; myeloid cells secrete VEGF in an efferocytosis-dependent manner [102]. Neovessels function as distribution lanes that provide oxygen and nutrients to metabolically active cells in the wounded area. During the inflammatory phase, neovessels are hyperpermeable and lack pericytes and thus facilitate leukocyte extravasation [61]. However, during the proliferative phase, mature neovessels are covered by a pericyte coat, which prohibits leukocyte extravasation and the angiogenic potential of ECs [61, 215, 294]. The formation of a pericyte coat requires platelet-derived growth factor (PDGF)-signalling and is mandatory for the formation of a mature scar [294]. A lack of the pericyte coat owing to the disruption of PDGF-signalling prolongs immune cell extravasation and decreases the collagen content and thus the stability of the mature scar [61].
Chronic phase after MI—the maturation phase
The key feature of a mature murine scar is an ECM that consists of cross-linked collagens, whereas the cellular effectors, such as MFs and ECs, have vanished.
Formation of a mature scar
The apoptosis of interstitial cells is induced by (1) the removal of matricellular proteins, (2) the deprivation of growth factors and (3) a reduced tensile force on MFs [215, 294]. The small proteoglycan biglycan orchestrates collagen organisation in the scar and an increased expression of lysil-oxydase catalyses the collagen cross-linking [55, 159, 273]. While collagen cross-linking increases the tensile strength of the myocardium and thereby may prevent chamber dilation, it can also induce diastolic dysfunction owing to increased cardiac stiffness. The formation of a supportive scar depends on the processes that take place during inflammation and proliferation: initial overshooting of inflammatory signals favours matrix degradation and thereby facilitates cardiac rupture during the acute phase [61]. Prolonged inflammation, however, delays the proliferative phase and thus impairs the deposition of fibrillar collagen, which may lead to ventricular dilation during the chronic phase after MI, owing to impaired tensile strength of the scar [61]. There are several differences between murine postischemic scars and those of larger mammals: (1) mice display extraordinary thin post-MI scars, due to lesser collagen in their scars [44]. (2) MFs do not exhibit a complete evanescence after scar formation in human MI but rather align parallel to the endocardium and the epicardium and thereby support the extracellular scar tissue [276]. Both may explain, why mice are highly prone to cardiac rupture after MI, whereas the phenomenon of postischemic rupture is rare in humans [121]. (3) myocardial scar formation is fast in mice compared to larger mammals as a mature scar is present within 14 days in murine I/R models, whereas scar formation in canine I/R protocols requires 28 days [43].
Interstitial remodelling upon myocardial infarction
Sufficient scar formation resolves proliferative signalling in the infarct zone; however, hemodynamically relevant MIs induce cardiac pressure and volume overload. This in turn increases neurohumoral activation and ventricular wall stretch [58] and thus induces cardiac hypertrophy and interstitial fibrosis in the primarily unaffected remote zone of the myocardium. Moreover, inadequately restricted cardiac inflammation extends the infiltration of immune cells into the remote zone. Here, these immune cells initiate interstitial inflammation and subsequent fibrosis without a mechanistic signification, which worsens cardiac remodelling and thus impairs cardiac function [61]. However, interstitial remodelling in the remote zone after MI in mice is less pronounced than in the prototypical murine models of hypertrophy and interstitial fibrosis, such as transverse aortic constriction [216].
Models of heart failure induced by pressure overload and neurohumoral activation
Transverse aortic constriction
Murine heart failure and its clinical relevance
Transverse aortic constriction (TAC) mimics increased left ventricular afterload in patients, which in the clinical setting predominantly occurs owing to aortic stenosis or systemic hypertension. TAC, and its clinical correlates, subject the left ventricle to pressure overload and subsequently activate neurohumoral stimuli. As a reaction to pressure overload and neurohumoral activation, cardiac hypertrophy and interstitial fibrosis occur. However, the acute onset of pressure overload in the murine model of TAC does not perfectly resemble the patients’ clinical course in which the pressure overload insidiously evolves over years or decades owing to aortic stenosis or hypertension. In mice, the model of TAC has been extensively used to examine signalling pathways that contribute to adverse cardiac remodelling and hypertrophy in the context of pressure overload and neurohumoral activation [112, 225, 252, 260]. As shown in Table 2, mice that are subjected to TAC exhibit cardiac remodelling in two sequential stages: (1) an initial stage of concentric hypertrophy and stable systolic but impaired diastolic function, thereby resembling features of human HF with preserved ejection fraction (HFpEF) [90, 252, 260]. (2) over time, the mice decompensate and exhibit severe systolic HF accompanied by eccentric hypertrophy, thereby resembling features of human HFrEF [90, 175, 252, 260]. However, the progression from HFpEF to HFrEF is not a mandatory sequela in the patients’ course of HFpEF [47].
Methodological proceedings
As depicted in Fig. 4a, TAC is performed by constricting the transverse aortic arch between the brachiocephalic trunk and the left carotid artery [225]. First, mice are anaesthetised and artificially ventilated. Hereafter, the chest is entered by an upper partial sternotomy and a suture is tied around the aortic arch against a cannula [225, 252]. The use of a cannula ensures that the aortic lumen is constricted to a defined diameter. By removing the cannula, the inner aortic diameter is reduced to about 0.4 mm, which constricts the aortic arch by approximately 70% [225]. A sufficient constriction establishes a pressure gradient across the stenosis of more than 40 mmHg [147]. Non-invasive pulse-wave doppler analysis can be used to measure this gradient and to subsequently select only animals with comparable gradients for further evaluation, which should be done to ensure a sound study [147, 157, 199]. However, the analysis of pressure gradients using a doppler ultrasound is limited to a maximum gradient of 60 mmHg across the stenosis [157]. Data on periprocedural mortality of TAC-surgeries range from less than 10% [225, 252] to more than 50% [11, 181, 249]. To induce a less severe TAC, some groups tie the suture against a thicker 26-gauge needle, by which a lower pressure gradient is established [27, 89].

Transverse aortic constriction (TAC) in mice. a Schematic drawing depicting the constriction of the transverse aortic arch between the brachiocephal trunk (1) and the other two branches, the left common carotid artery (2) and the subclavian artery (3). After tightening the suture against a 26- or 27-gauge cannula, the latter is removed, which leaves a defined aortic lumen behind. b Representative images of biventricular hearts (without atria) of mice that were either assigned to sham-surgery or to transverse aortic constriction (TAC) for 6 weeks demonstrate the enlarged ventricular mass observed after TAC in mice
Limitations
TAC-surgeries possess high procedural variability, depending on (1) the used mouse strain [13, 71], (2) the used sex [277], (3) the age at surgery and (4) the severity of the constriction [175, 260]. Thus, the increase in left ventricular mass varies highly even within the same strain; in one study the increase in left ventricular mass ranges from + 42% to + 376% after 8–9 weeks [175]. Figure 4b shows a hypertrophic murine heart, whose ventricular mass is enlarged by approx. 200% 6 weeks after TAC. In addition, the temporal dynamics of the transition from HFpEF to HFrEF vary even within the same experimental group [90]. Further, in a recent study only a subset of mice (28%) that were subjected to TAC develop signs of HFrEF [181], thus indicating that some mice will not proceed from HFpEF to HFrEF after TAC at all. Therefore, the authors suggest to examine cardiac function by means of echocardiography prior to further investigations and select a subgroup of animals, whose cardiac function indicates that they refer to the scope of investigation [90].
Recent advancements
In an attempt to reduce procedural variability, Melleby et al. recently introduced o-ring aortic banding with fixed diameter constrictors [175]. In their study, utilising o-rings with an inner diameter of 0.66 mm induces progressive concentric hypertrophy and interstitial fibrosis accompanied by left ventricular stiffness but stable contractile function (HFpEF). However, when subjected to TAC utilising o-rings with an inner diameter of 0.61 mm the mice showed early progression from concentric hypertrophy to dilated congestive HF (HFrEF) [175].
Chronic subjection to angiotensin II
Murine heart failure and its clinical relevance
As depicted in Fig. 5, chronic subjection to angiotensin II (Ang II) in mice mimics chronic hypertension due to neurohumoral activation as seen in HF patients. Neurohumoral activation includes the activation of the renin–angiotensin–aldosterone-system (RAAS), which results in elevated plasma levels of its main effector hormones Ang II and aldosterone. Thus, in spite of that it may often be seen as a hypertension model, chronic Ang II infusion resembles the myocardial injury and the subsequent cardiac remodelling owing to both, (1) cardiac pressure overload and (2) direct cardiac effects of Ang II and aldosterone. However, high Ang II levels that are observed in failing hearts do not only result from the activation of the systemic RAAS, but also from the activation of an intracardiac RAAS [205]. It has been postulated that the intracardiac cleavage of Ang I to Ang II in a local chymase-dependent manner is responsible for more than 75% of Ang II in failing hearts [133, 205]. Thus, it remains questionable, whether the infusion of supraphysiological doses of Ang II truly reflects the patients’ clinical setting, in which an already failing heart is confronted with Ang II from both, local and systemic, origin [260, 272].

Reciprocal interactions between pressure overload, RAAS activation, and heart failure. Simplified schematic drawing that visualises the reciprocal interactions between pressure overload, activation of the renin–angiotensin–aldosterone-system (RAAS), and heart failure (HF) in men and mice. The murine model of TAC mimics aortic stenosis in patients, in which cardiac pressure overload precedes the activation of the RAAS. The models of DOCA- or Ang II infusion, however, mimic the clinical course of patients, in which the development of HF, independent from its etiology, precedes the activation of the RAAS. However, sustained activation of the RAAS induces hypertension, thus RAAS activation also induces cardiac pressure overload. This initiates a self-perpetuating vicious circle of HF, RAAS activation and pressure overload. Further, since all here described murine models induce RAAS activation and pressure overload, the revelation of a distinct injurious stimuli in the development of HF and cardiac inflammation and fibrosis in these models is hampered
As shown in Table 2, mice subjected to Ang II infusion display cardiac remodelling in the presence [16, 271] as well as in the absence [214] of concomitant hypertension. This highlights that remodelling pathways exist that are independent from pressure overload. Depending on the dose and the time frame of subjection to Ang II, mice either exhibit concentric hypertrophy accompanied by diastolic dysfunction and thus resemble features of human HFpEF [17, 214], or they exhibit dilative, congestive HF accompanied by reduced ejection fraction and thus resemble features of human HFrEF [17, 271]. Likewise, the degree of hypertension depends on the dose and the time frame that mice are subjected to Ang II. Gomolak et al. analysed this dose-dependence in a study in which low Ang II-doses (approx. 0.1 mg/kg per day) do not induce hypertension, while intermediate doses (approx. 0.5 mg/kg per day) increase blood pressure over time and high doses (approx. 1.4 mg/kg per day) induce immediate and severe hypertension [78].
Methodological proceeding
Methodologically, osmotic minipumps, which subsequently release Ang II at a defined rate, are implanted subcutaneously [240]. Anaesthesia is maintained during surgery. However, no artificial ventilation is needed during the procedure, thereby making it less difficult than, e.g. the induction of MI or TAC. Typically, mice are subjected to Ang II for a time frame of 2–8 weeks [16, 17, 206, 240]. Sham controls are treated with volume-matched saline-vehicles. Though it would enhance reproducibility, blood levels of circulating Ang II are most often not measured [260].
Limitations
Even at a given infusion rate (1.4 mg/kg per day) and at a given time of subjection to Ang II (8 weeks), the degree of cardiac remodelling and dysfunction strongly depends on the specific mouse strain: while C57BL/6 mice in this context display concentric hypertrophy and preserved contractile function, Balb/c mice display congestive HF [206]. Strikingly, this is independent from the degree of hypertension, as both strains exhibit comparable blood pressures upon Ang II infusion [206]. This may in part be attributable to the predominantly Th1-mediated immune responses that are observed in C57BL/6 mice, whereas Balb/c mice predominantly exhibit Th2-mediated immune responses [206].
Chronic subjection to deoxycorticosterone acetate
Murine heart failure and its clinical relevance
As shown in Fig. 5, chronic subjection to the aldosterone analogue deoxycorticosterone acetate accompanied by unilateral nephrectomy and high salt diet (DOCA) promotes hypertension and the subsequent development of HF in mice [162, 244]. In the clinical setting, the combination of hypertension and elevated aldosterone levels promotes hypertensive heart disease and its transition to HFpEF, which is present in up to 50% of HF patients [98, 180, 201]. Strikingly, mice subjected to DOCA display mild hypertension and impairment of diastolic function accompanied by exercise intolerance but do not show signs of systolic dysfunction [118, 162, 260]. Thus, they are seen as one of the few murine models that truly resemble the features of human HFpEF [118, 162, 260]. In DOCA models, hypertension is known to appear in two phases, an initial peak in blood pressure, which is followed by sustained hypertension [14].
Methodological proceedings
Animals are anaesthetised and hereafter a unilateral nephrectomy is performed [162, 244, 260]. After the procedure, drinking water that contains 1% sodium chloride is applied. Few days after nephrectomy, pellets that release the aldosterone analogue DOCA at a constant rate are implanted subcutaneously [162, 244]. The development of diastolic dysfunction depends on the onset of hypertension, as mice that are solely subjected to DOCA without unilateral nephrectomy and high salt diet do not exhibit hypertension or impaired diastolic function [244].
Cardiac inflammation and fibrosis induced by pressure overload and neurohumoral activation
The common feature of the murine models of TAC or the subjection to Ang II or DOCA (in the following summarised as models of nonischemic HF) is the induction of HF and reactive interstitial and perivascular fibrosis upon pressure overload and neurohumoral activation. Neurohumoral systems, such as the sympathetic nervous system (SNS) and the renin–angiotensin–aldosterone-system (RAAS) are activated by low cardiac output to maintain hemodynamic homeostasis. However, low cardiac output is a feature of HF, thus HF itself leads to chronic and sustained activation of the SNS and the RAAS (Fig. 5). In a vicious circle, this initially adaptive response induces self-perpetuating cardiac remodelling and subsequent HF [65]. Table 2 provides an overview of the distinct characteristics of the murine models of nonischemic HF, such as (1) TAC in the late remodelling phase, (2) TAC in the early remodelling phase, (3) Ang II administered at high dose, (4) Ang II administered at low dose, (5) DOCA accompanied by concomitant unilateral nephrectomy and high salt diet, as well as (6) DOCA alone.
The initiation of inflammation and fibrosis in nonischemic as compared to ischemic models of heart failure
The development of interstitial fibrosis in the models of nonischemic HF is in contrast to the induction of reparative fibrosis in the models of ischemic HF, such as permanent MI or I/R: here, reparative fibrosis is the mechanistically rational process of wound healing; the circumscribed area of the unrecoverable infarct wall is replaced by fibrotic scar tissue to maintain cardiac structure. Thus, there is an immense disparity in the pathogenesis of cardiac inflammation and fibrosis in models of nonischemic and ischemic HF: (1) in ischemic HF, inflammation typically occurs prior to fibrosis in a sequential manner. This is in contrast to models of nonischemic HF, in which inflammation and fibrosis typically coexist. (2) in models of ischemic HF, the initial pro-inflammatory stimulus is precisely elaborated: necrotic cell death upon ischemia triggers the subsequent release of danger-signals, which in turn immediately alert the fire brigade of resident sentinel cells. Opposed to this, the significance of distinct pro-inflammatory stimuli in models of nonischemic HF is less elaborated: (i) pressure overload increases myocardial stretch and thereby induces pro-inflammatory cascades [153], (ii) pressure overload may induce a limited amount of cell death and may therefore induce the release of danger-signals, and (iii) the concomitant activation (and/or the preceding external infusion) of neurohumoral stimuli directly promotes fibrotic reprogramming in several cardiac cells and recruits monocytes from stem cell niches in spleen and bone marrow [26]. (3) the resolution of inflammation is precisely elaborated in the murine MI model: immune cells and CFs efferocyte dead cells and phagocyte danger signals; subsequently, they undergo a reparative reprogramming and thus provide a proliferative milieu. In models of nonischemic HF, however, interstitial fibrosis is a chronic and progressive epiphenomenon of the sustained repression of non-circumscribed, self-perpetuating inflammation and the concomitant chronic activation of pro-fibrotic stimuli.
Reciprocal interactions between pressure overload, RAAS activation and heart failure
As depicted in Fig. 5, the sequential occurrence of pressure overload and neurohumoral activation differs in the models of nonischemic HF: since pressure overload markedly activates the RAAS, pressure overload precedes RAAS activation in the TAC model [23]. In contrast, in the murine models of chronic administration of either Ang II or DOCA, RAAS-activation is mimicked, which precedes the induction of hypertension and thus pressure overload. However, as illustrated in Fig. 5, reciprocal interactions between pressure overload, RAAS activation, and heart failure exist; this hampers the revelation of the specific contribution of a distinct stimulus for the induction of inflammation and fibrosis in the models of TAC, Ang II and DOCA. Therefore, the pathogenesis of inflammation and fibrosis in these models is less understood [133].
The renin–angiotensin–aldosterone-system
As depicted in Fig. 6, the renin–angiotensin–aldosterone-system (RAAS) modulates vasotonus, renal water elimination, salt appetite, and the sympathetic activity of the central nervous system to ensure adequate organ perfusion. The RAAS consists of the interplay between distinct endocrine peptides. The liver secretes angiotensinogen, which is subsequently cleaved by the kidney-derived renin. Upon cleavage, angiotensinogen becomes the decapeptide angiotensin I (Ang I). Hereafter, Ang I is shortened to the octapeptide Ang II by the angiotensin-converting-enzyme (ACE), a membrane-bound metalloproteinase that is predominantly expressed on ECs of pulmonary vessels [205]. However, Ang I within the cardiac tissue is not only cleaved by ACE, but also by local, intracardiac chymases; a significant one is mast cell chymase [205]. In fact, more than 75% of the Ang II concentration in failing hearts may originate from a local RAAS [133].

The initiation of cardiac inflammation and fibrosis upon activation of the RAAS. The precursor of the renin–angiotensin–aldosterone-system (RAAS), angiotensinogen, is released by the liver; subsequently it is cleaved by the kidney-derived renin to angiotensin I, which is hereafter cleaved by the angiotensin-converting enzyme (ACE) or by intracardiac chymases, such as mast cell chymase, to form angiotensin II (Ang II). The latter evokes the release of aldosterone from the adrenal glands. Ang II and aldosterone (or its analogue DOCA) promote cardiac inflammation and fibrosis through the activation of systemic and intracardiac pathways. They provoke (1) salt appetite, sympathoexcitation, and the release of vasopressin in the brain, (2) increased vascular resistance, and (3) hypervolemia and hypernatremia through actions in the kidney and thereby induce hypertension. Subsequently, hypertension results in increased mechanical stretch on cardiac cells, which initiates cardiac remodelling through stretch-dependent signalling, especially in cardiac fibroblasts (CFs) and cardiomyocytes (CMs). Further, Ang II and aldosterone (or DOCA) directly initiate cardiac inflammation and fibrosis through actions in CFs, CMs, immune cells (ICs) and endothelial cells (ECs)
As depicted in Fig. 6, Ang II activates the angiotensin receptor type 1 (AT1R) pathway in the kidneys, the adrenal glands, the brain, the vessels, and directly in cells of the cardiac tissue and thereby mediates its adverse cardiovascular effects. (1) AT1R-signalling contracts vascular smooth muscle cells, which increases the peripheral resistance. (2) Ang II induces vascular remodelling, which results in endothelial dysfunction and further increased peripheral resistance. (3) Through actions in the circumventricular organs of the brain, such as the area postrema and the anteroventral third ventricle, Ang II alters the drinking behaviour, promotes sympathetic activation, and induces vasopressin release from the pituitary gland [84, 172]. Vasopressin is a potent vasoconstrictor and decreases the renal water clearance. (4) Ang II induces the release of the mineralocorticoid aldosterone from the adrenal gland. Aldosterone, and its analogue DOCA, are lipophilic molecules, which bind to their intracellular mineralocorticoid receptor (MR). In the kidneys, activation of the MR leads to increased retention of water and sodium and thereby induces hypervolemia. In the brain, activation of the MR promotes salt appetite and thus increases plasma osmolality [74, 84]. In a vicious circle, the circumventricular organs in turn detect the increased plasma osmolality and subsequently accentuate their sympathoexcitation and the release of vasopressin [84]. This further aggravates hypertension. The fact that lesions of the anteroventral third ventricle almost abolish a hypertensive phenotype in mice highlights the significance of the brain for the development of hypertension upon Ang II treatment [172]. Moreover, a recent study concludes that pro-inflammatory T-cell populations are essential for the development of hypertension upon neurohumoral activation [85]. As illustrated in Fig. 6, Ang II and aldosterone/DOCA directly mediate cardiac inflammation and fibrosis via actions on cells of the myocardium.
The direct impact of angiotensin II on cardiac cells
Pro-inflammatory Ang II-signalling
Ang II is a potent pro-inflammatory and pro-fibrotic hormonal mediator [272]. The downstream cascade of the G protein-coupled AT1R includes the activation of janus-kinase and signal transducers and activators of transcription (JAK-STAT) pathways, which ultimately result in the activation of transcription factors from the AP1-family and NFκB [40]; NFκB activation in this context further requires the presence of ROS [248]. However, Ang II itself induces this generation of ROS [125, 133]. In line with this, mice that overexpress a mitochondria-targeted catalase are protected from cardiac fibrosis after Ang II infusion [37]. If stimulated by Ang II, CMs and CFs release TNF-α and IL-6 [143, 283]. IL-6 may be a specific player of the induction of cardiac fibrosis in the context of Ang II infusion in mice, as the disruption of IL-6 prevents cardiac fibrosis in spite of not affecting hypertension or cardiac hypertrophy in this model [79]. IL-6 further orchestrates the expression of cytokines, chemokines and CAMs and thus promotes the activation of resident interstitial cells and the recruitment of immune cells. One example is the CCL-2 dependent recruitment of CD34+ CD45+ monocytic fibroblast precursors [46, 87]. These precursors mature into collagen-secreting fibroblasts in a TNF-receptor (TNFR)-1-dependent manner [46, 87]. In CFs, TNFR1-signalling upregulates AT1R expression and thereby sensitises these cells to Ang II stimulation [83]. Interestingly, Ang II or TNF-α alone do not induce the maturation of fibroblast precursors [46]. This indicates that the Ang II-mediated induction of fibroblast reprogramming under nonischemic conditions depends on initial inflammatory co-stimuli. Additionally, enhancer regions of MMP and TIMP genes possess AP-1 and NFκB binding sites [40], which promotes matrix remodelling upon Ang II stimulation. Further, circulating Ang II mediates the recruitment of the later-invading monocytes from their splenic reservoir [144].
Pro-fibrotic Ang II-signalling
The development of fibrosis upon Ang II infusion has two origins: (1) it is a sequela of the suppression of diffuse inflammation on the cost of activating TGF-β1-dependent, pro-fibrotic cascades. (2) Ang II directly promotes proliferative signalling especially in ECs, CFs and CMs. In these cells, it induces the release of growth factors such as PDGF, and TGF-β1, and the secretion of matricellular proteins, such as CCN-2 [143]. Like in other cardiac pathologies, TGF-β1 is one of the main conductors of fibrotic remodelling in the context of Ang II infusion. It was shown, that the upregulation of TGF-β1 through AT1R-signalling is mandatory for the development of cardiac fibrosis upon Ang II stimulation in mice [237]. Furthermore, the endothelial release of endothelin (ET)-1 upon Ang II stimulation is necessary for the development of HF in this model [2]. ET-1 enhances proliferation and attenuates apoptosis in CFs [133]. Moreover, ET-1 promotes matrix expansion as it stimulates collagen deposition and downregulates collagenase activity [133]. However, to selectively target pro-fibrotic pathways to prevent cardiac fibrosis is detrimental in the model of Ang II infusion, as it increases the susceptibility to cardiac rupture due to unrestricted inflammation [234]. However, the pro-fibrotic effects of cardiac AT1R-signalling may be fine-tuned by the anti-fibrotic effects of cardiac AT2R-signalling. In line with this, the CM-specific overexpression of AT2R decreases the degree of cardiac fibrosis but does not affect hypertension or cardiac hypertrophy in the context of chronic Ang II infusion in mice [139].
The direct impact of aldosterone/DOCA on cardiac cells
Aldosterone, and its analogue DOCA, directly mediate cardiac inflammation and fibrosis as they bind to their MRs in CMs, CFs, MPs, ECs and smooth muscle cells [133]. After nuclear translocation, MRs enhance the expression of proteins that are associated with cardiac inflammation and fibrosis, such as MMPs, TIMPs, TGF-β1, plasminogen activator inhibitor (PAI)-1, CCN-2, fibronectin and the collagens I, III, and IV [40, 255]. Furthermore, there is a growing body of evidence that various non-canonical pathways contribute to the transdifferentiation of MFs upon MR-signalling: both, MR-dependent ROS-generation and MR-dependent transactivation of several growth factor receptors, such as epidermal growth factor receptor (EGFR) and PDGF receptor (PDGFR), were shown to directly promote MF transdifferentiation [255].
MR-signalling in macrophages
On a cellular level, MPs seem to be crucially affected by MR-signalling in the murine models of nonischemic HF as MR-signalling critically polarises them towards a pro-inflammatory M1-phenotype [219, 259]. In line with this, MR-deficient MPs rather differentiate into M2-polarised MPs in a murine model of nonischemic HF [239]. Further, pro-inflammatory c-Jun NH2-terminal kinase/activating protein-1 (JNK/AP-1) and NFκB transactivation pathways depend on MR-signalling [239, 255]. However, it remains unclear, whether MRs in MPs solely promote their M1-polarisation, or if MRs in MPs are also mandatory for their recruitment: in a DOCA model, MP-specific MR-deficient mice are not protected from cardiac MP recruitment, but from hypertension [219]. In contrast, in a murine model that combines the infusion of the nitric oxide synthase inhibitor L-NAME with Ang II infusion, MP-specific MR-deficient mice are protected from cardiac MP recruitment, but not from hypertension [259]. However, MP-specific MR-deficient mice show less inflammation and less fibrosis in both models [219, 259].
MR-signalling in cardiomyocytes
CM-dependent MR-signalling additionally contributes to cardiac inflammation and fibrosis: depletion of CM-specific MR-signalling does not prevent immune cell infiltration but results in lesser collagen deposition in a murine DOCA model [217]. Interestingly, CM-specific MR-deficient mice show increased expression of decorin, an inhibitor of the pro-fibrotic mediators TGF-β1 and CCN-2; they further show increased MMP-2 and MMP-9 activity [217]. Taken together, the loss of CM-specific MR-signalling may not prevent immune cell recruitment, but may disrupt a pro-fibrotic vicious circle as it modulates TGF-β/CCN-2- and MMP-2/MMP-9-dependent pathways. In contrast, in models of TAC, CM-specific MR-depletion does not prevent cardiac inflammation and fibrosis [160].
MR-signalling in cardiac fibroblasts
CFs may also contribute to cardiac remodelling in a MR-dependent manner, as aldosterone in vitro stimulates the proliferation of CFs and increases their collagen synthesis [133]. Moreover, ECs express cytokines and CAMs, such as VCAM-1, in a MR-dependent manner [161]. In line with this, cardiac inflammation and fibrosis are less pronounced in EC-specific MR-deficient mice that are challenged with DOCA [161, 218]. However, as with the CM-specific MR-depletion, the EC-specific MR-depletion does not affect cardiac remodelling in murine TAC models [228].
Adrenergic signalling
Increased contractile demands of the myocardium and the concomitant upregulation of RAAS hormones in the models of TAC, Ang II, and DOCA activate the sympathetic nervous system (SNS) and thus promote the release of catecholamines. Moreover, sustained activity of the SNS is a key feature of human HF, hence β-blockade is a pivotal basis of its therapy [72]. The two major sources of catecholamines are (1) the adrenal glands, which predominantly release epinephrin upon sympathetic stimulation and (2) cardiac adrenergic nerve endings, which release norepinephrin [164]. On the one hand, activation of the SNS promotes fibrotic cardiac remodelling as it evokes the release of renin from the kidneys [24] and thus contributes to further RAAS-activation and hypertension. On the other hand, catecholamines directly promote adverse cardiac remodelling, as they bind to the G protein-coupled β-adrenergic receptors (β-AR)-1 and -2 on cardiac cells [164]. Recently, the complex downstream signalling of these receptors in the context of HF has been comprehensively reviewed [38].
There are three types of β-ARs, of which the β1-AR is predominantly expressed in healthy cardiac tissue (75–80%), followed by the β2- (15–20%) and the β3-AR (2-5%) [164]. However, chronic activation of the SNS upon HF in mice and men downregulates the β1- and desensitises the β1- and the β2-AR [164]. In turn, adrenergic signalling upon HF upregulates the G protein-coupled receptor kinase (GRK)-2, an enzyme that is known to promote fibrotic remodelling in experimentally induced HF [56]. Further, chronic activation of the β–AR-pathway in CMs directly induces hypertrophy and apoptosis and activates the transcription of a fetal gene programme [169]; further, it upregulates the myocardial expression of the pro-inflammatory cytokines IL-1β, -6, -18 and TNF-α in a NFκB-dependent manner [25, 191]. Upon chronic adrenergic stimulation, TNFR-1-signalling worsens cardiac remodelling, whereas TNFR-2-signalling may be cardioprotective [72]. Moreover, a recent study concludes that excessive CM-specific β1-AR-signalling generates ROS and thereby activates the inflammosome, which is required for the release of IL-18 [281]. However, incubating CFs with the supernatant of hypertrophic CMs, previously stressed by adrenergic stimulation, induces MF transdifferentiation and the subsequent secretion of collagens [91]. This paracrine effect is attributed to the ROS-dependent secretion of growth factors by CMs, such as CCN-2 [91]. Thus, a cross talk between CMs and CFs may be of overwhelming importance in the context of β-AR-mediated MF transdifferentiation. In contrast to the adverse effects of β1- and β2-AR-signalling, α1- (subtype A) [104, 198] and β3-AR-signalling [91] may have antipathetic effects as they ameliorate adverse remodelling and the transition to HFrEF upon pressure overload.
Mechanical stretch upon pressure overload and hypertension
Increased hemodynamic load after TAC, or to a minor degree upon Ang II- or DOCA-induced hypertension, increases the mechanical stretch on myocardial cells.
Cardiac fibroblasts—the sentinel cells that initiate inflammation upon mechanical stretch
Resident CFs are connected to the ECM through their transmembrane-spanning integrins; thus, they may be the sentinel cells that detect increased mechanical stretch [40, 153, 192, 280]. Mechanical stretch activates the downstream extracellular signal-regulated kinase (ERK), p38 and NFκB; subsequently, CFs secrete pro-inflammatory cytokines and chemokines of the CC-family, such as CCL-2, CCL-7, and CCL-8 [153]. Strikingly, chemokine secretion is not observed in CMs or ECs upon mechanical stretch [153]. Beyond that, mechanical stretch leads to sustained induction of the pro-inflammatory proteases MMP-2, MMP-9 and the CAM Cx43 [254]. In reaction to pro-inflammatory signals, CFs and immune cells further increase their MMP activity, which enables leukocytes to dig through the basal membrane and infiltrate the ECM [275]. This aggravates cardiac inflammation, as ECM fragments (matrikines) amplify the vicious circle of inflammation and leukocyte invasion [153, 221, 247, 280]. Interruption of this vicious circle of inflammation requires the activation of pro-fibrotic stimuli. Thus, inflammatory proteases, such as MMPs, activate TGF-β1 from latent stores; TGF-β1 is the key stimulus by which inflammatory CFs reprogramme to a matrix synthesising phenotype [143, 153]. In contrast to models of ischemic HF, in models of pressure overload only few of the activated, collagen-secreting CFs are α-SMA-positive MFs [185]. This highlights that CFs may not only follow one line of action in the murine models of HF, but possess a variety of reprogramming phenotypes that depend on the underlying pathophysiology. Though bone marrow-derived fibrocytes and endothelial-to-mesenchymal transition have long been under debate to significantly contribute to the population of activated CFs in pressure overloaded hearts, recent evidence suggests that activated CFs in this setting arise from a lineage of resident endothelial and epicardial fibroblasts [185]. Additionally, mechanical stretch directly upregulates TGF-β1 [153] and the matricellular protein CCN-2 [254]. Strikingly, CCN-2 inhibition does not affect cardiac remodelling in Ang II-infused mice, but prevents adverse left ventricular remodelling and cardiac dysfunction in mice subjected to TAC [143, 250].
Cardiomyocytes
Mechanical stretch induces CM hypertrophy and activates an intracardiac RAAS in CMs, which results in the myocardial accumulation of Ang II [56, 205]. Moreover, mechanical stretch in CMs promotes AT1R-signalling in a ligand-independent manner [293]. Beyond that, generation of oxidative stress in CMs upon pressure overload induces MR translocation in a ligand-independent manner [255]. Further, a limited amount of necrotic cell death may occur upon TAC [26]; the subsequent release of DAMPs induces an immediate inflammatory response through TLR-dependent pathways [133].
The transition from HFpEF to HFrEF in models of pressure overload
The development of compensatory concentric hypertrophy upon pressure overload initially is a response to the increased contractile demands of the myocardium. As a consequence, the increased stiffness of the hypertrophied myocardium leads to diastolic dysfunction and exercise intolerance (HFpEF). However, the subsequent transition to eccentric hypertrophy accompanied by severe systolic dysfunction (HFrEF) upon pressure overload is a pure pathologic sequela of self-perpetuating inflammation and fibrosis. Thus, the players that promote this transition are of great interest as they may depict key targets for clinical translation. Recent findings in a murine TAC model reveal that the early recruitment of monocytes is a key event in this context: pressure overload increases the number of CCR-2+ monocytes and their subsequent cardiac recruitment in the stage of HFpEF [204]. Hereafter, these cells differentiate into MPs and mediate the cardiac invasion of T-cells by presenting cardiac antigens on their surfaces [204]. This is of eminent importance, as the activation of CD4+ T-cells was shown to (1) depend on antigen presentation [122] and (2) to be pivotal for the transition from HFpEF to HFrEF in the context of pressure overload [142]; thus, the CCR-2-dependent recruitment of monocytes may be seen as an upstream mechanism of chronic inflammation that crosslinks innate and adaptive immunity, which ultimately results in HFrEF. On a molecular level, a tightly regulated, reciprocal interplay between matrix-degrading MMPs and matrix-preserving TIMPs may be responsible for the transition from HFpEF to HFrEF [93, 133]. In HFpEF, MMPs remain rather quiescent, whilst they are active in HFrEF [113]. In contrast, pro-fibrotic stimuli such as TGF-β1 remain rather quiescent in HFrEF [133].
Vascular remodelling upon pressure overload
Pressure overload in mice induces cardiac angiogenesis and extensive vascular remodelling [174, 280]. As pressure overload increases the blood pressure in coronary arteries, perivascular fibrosis may depict a beneficial response to resist these higher perfusion pressures [132]. Furthermore, in pressure overload models coronary arterioles promote myocardial fibrosis as they develop adventitial inflammation in an Ang II-dependent manner; inflammation subsequently extends and promotes perivascular fibrosis [173, 280]. However, vascular cells additionally possess anti-fibrotic features: ECs secrete interferon-γ-inducible protein (IP)-10/CXCL10 and hypoxia-inducible factor (HIF)-1; both of them suppress TGF-β-signalling [133].
Model of heart failure induced by viral myocarditis
Murine heart failure model and its clinical relevance
Myocarditis is an inflammatory disease of the myocardium that is predominantly caused by infectious agents [275]. The clinical course of myocarditis has a broad spectrum of outcomes, ranging from complete recovery to dilated cardiomyopathy (DCM) in the context of viral elimination or chronic inflammatory cardiomyopathy in the context of viral persistence. About 10% of patients with biopsy-proven viral myocarditis develop inflammatory cardiomyopathy [7]. However, myocarditis may remain asymptomatic and thus displays a significant cause of sudden death that occurs especially in young adults [34, 50, 236].
Depending on their susceptibility to the virus, mice, likewise humans, exhibit a variety of outcomes after infection with Coxsackievirus B3 (CVB3) [129, 151]. All strains, susceptible (e.g. BALB/c) or not (e.g. C57BL/6), initially suffer from acute myocarditis as characterised by systolic and diastolic dysfunction alongside interstitial remodelling [115, 129, 267]. Opposed to this, solely susceptible strains develop chronic myocarditis, also described as inflammatory cardiomyopathy, as a result of virus persistence or non-resolved cardiac inflammation. This is characterised by cardiac inflammation, reactive fibrosis alongside deteriorated cardiac function. Inoculating mice from susceptible strains with CVB3 therefore resembles features of inflammatory cardiomyopathy in humans.
Methodological proceedings
Coxsackievirus B3 is a non-enveloped, single-stranded, positive-sense RNA virus within the family of the picornaviredae. Commonly, 104–107 plaque-forming units of the heart passaged enterovirus CVB3 are injected intraperitoneally [15, 16, 129, 203]. Several CVB3 viral strains exist; a frequently used one is the Nancy-strain [123, 176]. The potency and the efficacy of a certain viral strain may vary, which should be considered in the experimental setup [8, 107]. As depicted in Fig. 7, the genome of CVB3 encodes 11 proteins, 4 structural proteins and 7 non-structural proteins. Since CVB3 is a positive-sense RNA virus, its RNA genome is directly translated into proteins upon host cell infection and hereafter functions as the template for viral replication. Appropriate viral infection can be proven by a loss of bodyweight and the detection of viremia in small serum samples, which typically occurs around day 2–4 post injection (p.i.). Tissue sampling and hemodynamic assessments should be performed either from day 6 to 8 during the acute phase of myocarditis or around day 28 during the chronic phase, depending on the curiosity for acute or chronic cardiac remodelling, respectively [15, 16, 150, 151, 274].

The course of viral myocarditis induced by CVB3 in mice. Inoculating mice with CVB3 results in viral myocarditis, which, likewise in humans, occurs in three sequential stages. The first stage, virus-mediated myocarditis, is observed from day 2 to 3 p.i. in mice. During this stage, the non-structural viral proteases 2A and 3C directly damage cells of the cardiac tissue and thereby initiate inflammation. Hereafter, sentinel cells are alarmed and recruit a first wave of immune cells into cardiac tissue, as shown in the representative hematoxylin and eosin (H&E) staining [15]. This results in the second stage, immune-mediated myocarditis, from day 4 to 14. During this phase, pro-inflammatory leukocytes either eliminate the virus (non-susceptible strains) or solely cease viral replication (susceptible strains); however, the leukocytes severely harm the cardiac tissue in both cases. In the case of viral elimination, cardiac remodelling is reversible and may either result in complete recovery or in dilated cardiomyopathy, depending on the degree of cardiac damage during acute myocarditis. In the case of viral persistence, a second wave of immune cells is recruited and mediates a chronic cardiac inflammation that is accompanied by dilated cardiomyopathy, known as inflammatory cardiomyopathy. Additionally, molecular mimicry may induce the development of a self-reactive autoimmunity in susceptible strains, which equally results in inflammatory cardiomyopathy
Cardiac inflammation and fibrosis induced by viral myocarditis
Upon inoculation with CVB3 in mice, myocarditis occurs in three sequential stages [66, 151]: (1) virus-mediated myocarditis from day 2 to 4, (2) immune-mediated myocarditis from day 4 to 14, and (3) the subsequent chronic stage of cardiac remodelling or chronic inflammation, which typically occurs around day 28. Virus-mediated myocarditis is characterised by viral entry and the translation of viral proteins in cardiac host cells. During this phase, the translated viral proteins themselves exert direct cytotoxic damage; however, infiltrating immune cells are absent [151]. The subsequent stage of immune-mediated myocarditis is characterised by a first wave of immune cell infiltration and the active replication of the virus in its host cells [151]. Activation of the host immunity eliminates the virus in non-susceptible strains or ceases viral replication in susceptible strains; however, this happens on the cost of initiating a thunderous storm of cardiac inflammation. The stages (1) and (2) are frequently summarised as acute viral myocarditis. The chronic stage in susceptible strains is characterised by a second wave of immune cell infiltration, which ultimately results in inflammatory cardiomyopathy. At this stage, viral replication has ceased, but the viral genome persists in cardiac tissue [151]. Beyond the genetic determination of the mouse strain, several other preconditions, such as age, nutrition status, gestation, and sex hormones were found to further determine the susceptibility to CVB3 infections in mice [284].
Virus-mediated acute myocarditis
CVB3 typically enters its host via the gastrointestinal or the respiratory tract [235], whereas it is inoculated intraperitoneally in the experimental setup. CVB3 enters CMs upon viremia 2–3 days after inoculation and subsequently exerts cell-to-cell spread [129, 235, 284]. Beyond that, in vitro evidence suggests a role for infected CFs [141, 151]. CVB3 enters CMs through interactions with the coxsackie-adenovirus receptor (CAR) [66, 129] and uses the decay acceleration factor (DAF) as a coreceptor [284]. In line with this, depleting the CAR in CMs [241] or treating CVB3-inoculated mice with soluble CARs [208] blunts acute myocarditis in this model. Higher CAR-expression in younger individuals may further determine their higher susceptibility to viral myocarditis [284]. After entering its host cell, the progeny viral RNA is translated and replicated and subsequently harms its host cells predominantly via two viral proteins, the proteases 2A and 3C. Both proteases are encoded within the viral genome and are immediately translated upon host cell infection. They cleave a variety of intracellular target proteins [66, 73]: (1) 2A and 3C break down protein biosynthesis by cleaving an initiation factor of translation (eIF-4G); (2) 2A and 3C activate caspase-8, by which apoptosis is induced; (3) 2A cleaves the transcription factor serum response factor (SRF) and thereby decreases the expression of contractile proteins; (4) 2A cleaves structural proteins such as dystrophin and dysferlin and thereby destabilises cellular and organellar membranes. This implies severe consequences for cardiac contraction as the dystrophin-cleavage affects the integrity of the sarcolemma membrane [10]. (5) 2A and 3C cleave autophagy-associated molecules. Finally, all these damages result in the lysis of host cells [73]. Strikingly, mice develop dilated cardiomyopathy after induced CM-restricted expression of the viral protease 2A in the absence of concomitant CVB3 infection [282]; further, they develop CVB3-induced myocarditis after the depletion of T- and B-cells [29]. Thus, host immunity is not required for the development of an initial virus-mediated cardiac injury. However, several resident cardiac cells, such as ECs, CMs, CFs and MPs are activated upon (1) viral entry into host cells and (2) virus-mediated cardiac injury and subsequently release pro-inflammatory mediators [275]. Thereby, they promote leukocyte invasion, fuel inflammation, and thus initiate the immune-mediated stage of viral myocarditis.
Immune-mediated acute myocarditis (antiviral immunity)
The fire brigade of innate immune cells invades the cardiac tissue 4–5 days p.i. and draws the virus into a fight for survival [103]. At this point, the initial virus-mediated myocardial inflammation proceeds to an immune-mediated one. Subsequently, cells of the adaptive immunity arrive 6–7 days p.i. to support the innate immune cells [284]. However, their invasion is associated with overshooting inflammation and subsequent adverse remodelling [284]. CM death and the infiltration of immune cells peak from day 9 to day 12 p.i., respectively [129]. The stage of immune-mediated acute myocarditis is critical for the long-term outcome, as viral elimination may permit reverse cardiac remodelling, whereas viral persistence results in sustained inflammation and non-reversible cardiac remodelling [284].
Activation of the innate immune system
Pattern recognition receptors (PRRs) of the innate immune system, such as TLR-3 [67] and TLR-7/8 [284] detect viral single-stranded RNA and intermediate viral double-stranded RNA in infected cells [73]. Sex differences in TLR-expression may explain the high susceptibility of male mice as compared to females [223, 224]. After CVB3 infection, TLR-3, -7, -8, and -9, all predominantly localised in endosomes, are upregulated in mice [16, 251]. Hence, they are of predominant importance after viral internalisation in phagocytes. However, upon viral replication in host cells, the double-stranded intermediate RNA predominantly accumulates in the cytosol [284]. Therefore, RNA helicases, such as retinoic acid-induced protein I (RIG-I) and melanoma differentiation-associated gene 5 (MDA-5), detect cytosolic dsRNA in host cells [124]. Once alarmed by pro-inflammatory cascades downstream of their TLRs, innate immune cells secrete antiviral type I interferones (IFNs), such as IFN-α and IFN-β [66]. Type I IFNs possess antiviral features and inhibit viral replication. Murine knock out studies revealed that in the context of CVB3-induced myocarditis the depletion of the type I IFN-receptor worsens mortality [269]; however, as viral replication in cardiac tissue remains unaffected, this may rather be an epiphenomenon of higher hepatic viral replication. Pivotal regulators of IFN-β are protease-activated receptor (PAR)-1 and PAR-2: while the activation of PAR-1 enhances IFN-β secretion, the activation of PAR-2 downregulates IFN-β expression [268]. Moreover, innate immune cells, but also CFs [151], secrete pro-inflammatory cytokines such as IL-1 and TNF-α; this amplifies cardiac inflammation and the recruitment of further immune cells [73]. IL-1 and TNF-α may display pro-inflammatory key stimuli, as prolonged and sustained secretion of IL-1 [149] and TNF-α [105] has both been shown to exaggerate viral myocarditis in mice. Recent evidence suggests that specifically neutralising IL-1β reduces cardiac inflammation and fibrosis and prevents chronic viral myocarditis in mice [135]. Further, the treatment with a soluble TNF-receptor [137] or a neutralising chemokine-antibody [274] lowers cardiac inflammation and protects the heart from myocardial injury. Moreover, the expression of a variety of MMPs is upregulated upon stimulation with pro-inflammatory cytokines [275]. The secretion of MMP-9 by invading immune cells is one example of how they break through the basal membrane [275]. Moreover, the collagen-turnover shows both qualitative and quantitative changes during acute myocarditis: beyond a tiny reduction in absolute cardiac collagen content, the fraction of soluble collagen is increased during acute myocarditis in mice [275]. A high fraction of soluble collagen is associated with impaired collagen cross-linking, which is known to promote adverse remodelling [279]. Further, soluble collagen itself may promote vascularisation of the injured tissue and may permit myocyte-slippage, a process thought to play a crucial role in the development of DCM even after complete viral elimination [275]. In line with this, pro-inflammatory chemokines (e.g. CCL-2, -5, and -7) and cytokines (e.g. IL-1, IL-6, and TNF-α) are upregulated alongside pro-fibrotic players (e.g. TGF-β1, TIMP-1, collagen-1 and -3, as well as CCN-2) during acute myocarditis [15]. Strikingly, the expression levels of the aforementioned proteins return to baseline after acute myocarditis in non-susceptible strains, whereas they remain elevated in susceptible strains.
Activation of the adaptive immune system
Once they engulfed viral particles and dead cells, MPs and dendritic cells present viral fragments on their surface and thereby recruit players of the adaptive immune system. However, T-cell recruitment is an ambiguous event in the context of viral myocarditis: on the one hand, T-cells are essential for the sufficient elimination of the virus, on the other hand, viral elimination includes the clearance of infected CMs. Since CMs possess negligible regenerative capacity in mammals, the T-cell mediated clearance of infected CMs (1) reduces the contractility of the myocardium and (2) induces cardiac remodelling to replace CMs with ECM. In line with this, T-cell infiltration is temporarily accompanied by pathological damage in CVB3-infected cardiac tissue [128]. Accordingly, the depletion of CD4+ and CD8+ T-cells has been shown to dampen mortality and inflammation in CVB3-induced myocarditis [200]. Strikingly, complete T-cell depletion does not affect cardiac viral titres [284]. This highlights the role of the innate immunity, which precedes the time-consuming activation of antigen-specific immunity, in limiting early replication and propagation of CVB3 [284]. However, excessive innate-mediated inflammation may favour the early recruitment of circulating fibroblast precursors; in the inflamed myocardium, these precursors transdifferentiate and, as contrarily described in ischemic HF, may exhibit a major source of MFs in the context of myocarditis-related cardiac remodelling [56]. This is in line with a recent study which shows that the depletion of MPs is associated with higher viral titres during acute myocarditis, but with fewer MFs and thus less fibrotic long-term remodelling in the chronic phase [117, 152].
The chronic stage of myocarditis (viral elimination or viral persistence and anti-self immunity)
Viral elimination
As depicted in Fig. 7, the chronic stage after CVB3-induced myocarditis is highly variable in mice. In non-susceptible strains, sufficient viral elimination may either result in complete recovery or lead to sterile DCM, depending on the severity of cardiac injury during acute myocarditis. Thus, after the first wave of immune cells has vanished, reversible cardiac remodelling is frequently observed in non-susceptible strains [15]. However, cardiac contractility may remain impaired despite reversed morphologic remodelling in non-susceptible strains [15]. One may speculate that this is observed, at least in part, due to the loss of CMs during acute myocarditis owing to (1) myocyte-slippage and (2) to the lytic cycle of the virus. Since myocardial fibrosis is reversible in this context, it is likely that collagen cross-linking does not appear during the acute stages of myocarditis in non-susceptible strains, as it would impair the collagenase-mediated collagen cleavage that is required for reverse cardiac remodelling [15, 227]. In line with this, an increased fraction of soluble collagen is observed during acute myocarditis [275].
Viral persistence
In stark contrast, in susceptible strains, a second wave of immune cells enters the cardiac tissue after the first wave has vanished [51] due to at least two different reasons, virus persistence or autoimmunity. Susceptible mouse strains can only hinder viral replication but do not completely eliminate the viral genome from their cardiac tissue. As these strains develop inflammatory dilated cardiomyopathy following acute viral myocarditis, it is likely that the persistence of the virus is, at least in part, responsible for such sequelae. In line with this, the CM-specific expression of a replication-incompetent CVB3 cDNA in mice leads to (non-replicating) viral persistence in CMs, which in turn results in inflammatory dilated cardiomyopathy [270, 284].
Due to the lysis of cells as the final step in the lytic cycle of viral replication, intracellular host proteins may induce the development of a self-reactive autoimmunity, which additionally contributes to the development of inflammatory dilated cardiomyopathy [66]. In this context, molecular mimicry initiates the autoimmune responses [226]; the exposure of sarcomeric fragments (e.g. myosin) that cross-react with CVB3-antigens on MHC molecules recruits self-reactive immune cells, which attack CMs through direct cytotoxicity or through the secretion of auto-antibodies [66]. As with viral persistence, the initiation of autoimmunity upon CVB3-infection is exclusively seen in susceptible mouse strains [51, 226].
Conclusion
Several murine models that develop HF accompanied by cardiac inflammation and fibrosis are frequently used in experimental setups. As summarised in Table 3, each model has features that makes it unique when compared to the other models. Thus, it may remain challenging to select an appropriate model that fits both, (1) the exploration of the scientific hypothesis in a setting with clinical relevance and (2) the technical capacity of the laboratory or its cooperatives.
While the ischemic models of HF and TAC require elaborate surgical skills owing to the need for artificial ventilation and access to the thorax, the subjection of mice to Ang II or CVB3 does not require this and is therefore less complex. As the DOCA regimen includes a unilateral nephrectomy but no access to the thorax, it may stand in between the aforementioned models regarding its feasibility. Further, the initial injurious stimuli and thus the subsequent development of HF, inflammation and fibrosis may be divided into two paramount categories in the here-described murine models of HF: On the one hand, ischemic models exhibit a local injury which predominantly initiates reparative fibrosis and results in HFrEF. This injury is transmural in the model of permanent ischemia but rather midmyocardial in the model of ischemia–reperfusion. On the other hand, nonischemic models exhibit a diffuse injury of the myocardium that subsequently initiates interstitial fibrosis and either results in HFpEF or HFrEF, depending on the severity and the duration of the injurious stimulus. Hypertrophy, however, is most pronounced in the models of TAC and Ang II, but may also be seen in the remote zone after MI. However, TAC is the model, which is preferentially used for the analysis of hypertrophy in mice. Concerning the initiation of inflammation, pattern recognition receptors (PRRs) play a pivotal role in the models of ischemic HF due to the release of danger-associated molecular patterns (DAMPs) upon CM-necrosis. However, PRRs additionally play a crucial role in the nonischemic model of viral myocarditis due to the pathogen-associated molecular patterns (PAMPs) that alarm sentinel cells and invading leukocytes. However, a limited amount of CM-necrosis and thus PRR-signalling may also occur in the murine models of TAC, Ang II and DOCA owing to cardiac pressure overload, which induces the release of DAMPs upon CM-necrosis or apoptosis. The prototypical model that mimics neurohumoral activation is Ang II; however, concomitant neurohumoral activation may additionally occur in all here-described models due to cardiac pressure overload and the development of HF (as shown in Fig. 5). Likewise, cardiac pressure overload is a prototypical characteristic of the TAC model, but may concomitantly occur in all other here-described models, either due to neurohumoral activation and subsequent hypertension, or due to impaired cardiac function.
References
Aartsen WM, Pelsers MM, Hermens WT, Glatz JF, Daemen MJ, Smits JF (2000) Heart fatty acid binding protein and cardiac troponin T plasma concentrations as markers for myocardial infarction after coronary artery ligation in mice. Pflugers Arch 439:416–422. https://doi.org/10.1007/s004249900180
Adiarto S, Heiden S, Vignon-Zellweger N, Nakayama K, Yagi K, Yanagisawa M, Emoto N (2012) ET-1 from endothelial cells is required for complete angiotensin II-induced cardiac fibrosis and hypertrophy. Life Sci 91:651–657. https://doi.org/10.1016/j.lfs.2012.02.006
Alard JE, Ortega-Gomez A, Wichapong K, Bongiovanni D, Horckmans M, Megens RT, Leoni G, Ferraro B, Rossaint J, Paulin N, Ng J, Ippel H, Suylen D, Hinkel R, Blanchet X, Gaillard F, D’Amico M, von Hundelshausen P, Zarbock A, Scheiermann C, Hackeng TM, Steffens S, Kupatt C, Nicolaes GA, Weber C, Soehnlein O (2015) Recruitment of classical monocytes can be inhibited by disturbing heteromers of neutrophil HNP1 and platelet CCL5. Sci Transl Med 7:317ra196. https://doi.org/10.1126/scitranslmed.aad5330
Alex L, Frangogiannis NG (2018) The cellular origin of activated fibroblasts in the infarcted and remodeling myocardium. Circ Res 122:540–542. https://doi.org/10.1161/CIRCRESAHA.118.312654
Andersson L, Scharin Tang M, Lundqvist A, Lindbom M, Mardani I, Fogelstrand P, Shahrouki P, Redfors B, Omerovic E, Levin M, Boren J, Levin MC (2015) Rip2 modifies VEGF-induced signalling and vascular permeability in myocardial ischaemia. Cardiovasc Res 107:478–486. https://doi.org/10.1093/cvr/cvv186
Annes JP, Munger JS, Rifkin DB (2003) Making sense of latent TGFbeta activation. J Cell Sci 116:217–224. https://doi.org/10.1242/jcs.00229
Anzini M, Merlo M, Sabbadini G, Barbati G, Finocchiaro G, Pinamonti B, Salvi A, Perkan A, Di Lenarda A, Bussani R, Bartunek J, Sinagra G (2013) Long-term evolution and prognostic stratification of biopsy-proven active myocarditis. Circulation 128:2384–2394. https://doi.org/10.1161/CIRCULATIONAHA.113.003092
Arola A, Kalimo H, Ruuskanen O, Hyypia T (1995) Experimental myocarditis induced by two different coxsackievirus B3 variants: aspects of pathogenesis and comparison of diagnostic methods. J Med Virol 47:251–259. https://doi.org/10.1002/jmv.189047031
Arslan F, Smeets MB, Riem Vis PW, Karper JC, Quax PH, Bongartz LG, Peters JH, Hoefer IE, Doevendans PA, Pasterkamp G, de Kleijn DP (2011) Lack of fibronectin-EDA promotes survival and prevents adverse remodeling and heart function deterioration after myocardial infarction. Circ Res 108:582–592. https://doi.org/10.1161/CIRCRESAHA.110.224428
Badorff C, Lee GH, Lamphear BJ, Martone ME, Campbell KP, Rhoads RE, Knowlton KU (1999) Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat Med 5:320–326. https://doi.org/10.1038/6543
Baicu CF, Zhang YH, Van Laer AO, Renaud L, Zile MR, Bradshaw AD (2012) Effects of the absence of procollagen C-endopeptidase enhancer-2 on myocardial collagen accumulation in chronic pressure overload. Am J Physiol Heart Circ Physiol 303:H234–H240. https://doi.org/10.1152/ajpheart.00227.2012
Barcellos-Hoff MH, Derynck R, Tsang ML, Weatherbee JA (1994) Transforming growth factor-beta activation in irradiated murine mammary gland. J Clin Investig 93:892–899. https://doi.org/10.1172/jci117045
Barrick CJ, Dong AP, Waikel R, Corn D, Yang F, Threadgill DW, Smyth SS (2009) Parent-of-origin effects on cardiac response to pressure overload in mice. Am J Physiol Heart Circ Physiol 297:H1003–H1009. https://doi.org/10.1152/ajpheart.00896.2008
Basting T, Lazartigues E (2017) DOCA-salt hypertension: an update. Curr Hypertens Rep 19:32. https://doi.org/10.1007/s11906-017-0731-4
Becher PM, Gotzhein F, Klingel K, Escher F, Blankenberg S, Westermann D, Lindner D (2017) Cardiac function remains impaired despite reversible cardiac remodeling after acute experimental viral myocarditis. J Immunol Res 2017:6590609. https://doi.org/10.1155/2017/6590609
Becher PM, Hinrichs S, Fluschnik N, Hennigs JK, Klingel K, Blankenberg S, Westermann D, Lindner D (2018) Role of Toll-like receptors and interferon regulatory factors in different experimental heart failure models of diverse etiology: IRF7 as novel cardiovascular stress-inducible factor. PLoS One 13:e0193844. https://doi.org/10.1371/journal.pone.0193844
Becher PM, Lindner D, Miteva K, Savvatis K, Zietsch C, Schmack B, Van Linthout S, Westermann D, Schultheiss HP, Tschope C (2012) Role of heart rate reduction in the prevention of experimental heart failure: comparison between If-channel blockade and beta-receptor blockade. Hypertension 59:949–957. https://doi.org/10.1161/HYPERTENSIONAHA.111.183913
Bhattacharya K, Farwell K, Huang M, Kempuraj D, Donelan J, Papaliodis D, Vasiadi M, Theoharides TC (2007) Mast cell deficient W/Wv mice have lower serum IL-6 and less cardiac tissue necrosis than their normal littermates following myocardial ischemia-reperfusion. Int J Immunopathol Pharmacol 20:69–74. https://doi.org/10.1177/039463200702000108
Bliksoen M, Mariero LH, Torp MK, Baysa A, Ytrehus K, Haugen F, Seljeflot I, Vaage J, Valen G, Stenslokken KO (2016) Extracellular mtDNA activates NF-kappaB via toll-like receptor 9 and induces cell death in cardiomyocytes. Basic Res Cardiol 111:42. https://doi.org/10.1007/s00395-016-0553-6
Brown LF, Dubin D, Lavigne L, Logan B, Dvorak HF, Van de Water L (1993) Macrophages and fibroblasts express embryonic fibronectins during cutaneous wound healing. Am J Pathol 142:793–801
Brown RD, Ambler SK, Mitchell MD, Long CS (2005) The cardiac fibroblast: therapeutic target in myocardial remodeling and failure. Annu Rev Pharmacol Toxicol 45:657–687. https://doi.org/10.1146/annurev.pharmtox.45.120403.095802
Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, Frangogiannis NG (2008) Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol 173:57–67. https://doi.org/10.2353/ajpath.2008.070974
Bujak M, Frangogiannis NG (2007) The role of TGF-beta signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res 74:184–195. https://doi.org/10.1016/j.cardiores.2006.10.002
Camacho Londono JE, Tian Q, Hammer K, Schroder L, Camacho Londono J, Reil JC, He T, Oberhofer M, Mannebach S, Mathar I, Philipp SE, Tabellion W, Schweda F, Dietrich A, Kaestner L, Laufs U, Birnbaumer L, Flockerzi V, Freichel M, Lipp P (2015) A background Ca2+ entry pathway mediated by TRPC1/TRPC4 is critical for development of pathological cardiac remodelling. Eur Heart J 36:2257–2266. https://doi.org/10.1093/eurheartj/ehv250
Chandrasekar B, Marelli-Berg FM, Tone M, Bysani S, Prabhu SD, Murray DR (2004) Beta-adrenergic stimulation induces interleukin-18 expression via beta2-AR, PI3 K, Akt, IKK, and NF-kappaB. Biochem Biophys Res Commun 319:304–311. https://doi.org/10.1016/j.bbrc.2004.04.185
Chen B, Frangogiannis NG (2018) The role of macrophages in nonischemic heart failure. JACC Basic Transl Sci 3:245–248. https://doi.org/10.1016/j.jacbts.2018.03.001
Chen H, Hwang H, McKee LA, Perez JN, Regan JA, Constantopoulos E, Lafleur B, Konhilas JP (2013) Temporal and morphological impact of pressure overload in transgenic FHC mice. Front Physiol 4:205. https://doi.org/10.3389/fphys.2013.00205
Chen W, Spitzl A, Mathes D, Nikolaev VO, Werner F, Weirather J, Spiranec K, Rock K, Fischer JW, Kammerer U, Stegner D, Baba HA, Hofmann U, Frantz S, Kuhn M (2016) Endothelial actions of ANP enhance myocardial inflammatory infiltration in the early phase after acute infarction. Circ Res 119:237–248. https://doi.org/10.1161/CIRCRESAHA.115.307196
Chow LH, Beisel KW, McManus BM (1992) Enteroviral infection of mice with severe combined immunodeficiency. Evidence for direct viral pathogenesis of myocardial injury. Lab Invest 66:24–31
Cimini M, Fazel S, Zhuo S, Xaymardan M, Fujii H, Weisel RD, Li RK (2007) c-kit dysfunction impairs myocardial healing after infarction. Circulation 116:I77–I82. https://doi.org/10.1161/CIRCULATIONAHA.107.708107
Cleutjens JP, Verluyten MJ, Smiths JF, Daemen MJ (1995) Collagen remodeling after myocardial infarction in the rat heart. Am J Pathol 147:325–338
Cochain C, Channon KM, Silvestre JS (2013) Angiogenesis in the infarcted myocardium. Antioxid Redox Signal 18:1100–1113. https://doi.org/10.1089/ars.2012.4849
Cohen M, Boiangiu C, Abidi M (2010) Therapy for ST-segment elevation myocardial infarction patients who present late or are ineligible for reperfusion therapy. J Am Coll Cardiol 55:1895–1906. https://doi.org/10.1016/j.jacc.2009.11.087
Cooper LT Jr (2009) Myocarditis. N Engl J Med 360:1526–1538. https://doi.org/10.1056/NEJMra0800028
Corbett SA, Schwarzbauer JE (1998) Fibronectin-fibrin cross-linking: a regulator of cell behavior. Trends Cardiovasc Med 8:357–362. https://doi.org/10.1016/S1050-1738(98)00028-0
Creemers E, Cleutjens J, Smits J, Heymans S, Moons L, Collen D, Daemen M, Carmeliet P (2000) Disruption of the plasminogen gene in mice abolishes wound healing after myocardial infarction. Am J Pathol 156:1865–1873. https://doi.org/10.1016/S0002-9440(10)65060-2
Dai DF, Johnson SC, Villarin JJ, Chin MT, Nieves-Cintron M, Chen T, Marcinek DJ, Dorn GW 2nd, Kang YJ, Prolla TA, Santana LF, Rabinovitch PS (2011) Mitochondrial oxidative stress mediates angiotensin II-induced cardiac hypertrophy and Galphaq overexpression-induced heart failure. Circ Res 108:837–846. https://doi.org/10.1161/CIRCRESAHA.110.232306
de Lucia C, Eguchi A, Koch WJ (2018) New insights in cardiac beta-adrenergic signaling during heart failure and aging. Front Pharmacol 9:904. https://doi.org/10.3389/fphar.2018.00904
DeLeon-Pennell KY, Iyer RP, Ero OK, Cates CA, Flynn ER, Cannon PL, Jung M, Shannon D, Garrett MR, Buchanan W, Hall ME, Ma Y, Lindsey ML (2017) Periodontal-induced chronic inflammation triggers macrophage secretion of Ccl12 to inhibit fibroblast-mediated cardiac wound healing. JCI Insight. https://doi.org/10.1172/jci.insight.94207
Deschamps AM, Spinale FG (2006) Pathways of matrix metalloproteinase induction in heart failure: bioactive molecules and transcriptional regulation. Cardiovasc Res 69:666–676. https://doi.org/10.1016/j.cardiores.2005.10.004
Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G (1993) Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol 122:103–111. https://doi.org/10.1083/jcb.122.1.103
Dewald O, Frangogiannis NG, Zoerlein MP, Duerr GD, Taffet G, Michael LH, Welz A, Entman ML (2004) A murine model of ischemic cardiomyopathy induced by repetitive ischemia and reperfusion. Thorac Cardiovasc Surg 52:305–311. https://doi.org/10.1055/s-2004-821153
Dewald O, Ren G, Duerr GD, Zoerlein M, Klemm C, Gersch C, Tincey S, Michael LH, Entman ML, Frangogiannis NG (2004) Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol 164:665–677. https://doi.org/10.1016/S0002-9440(10)63154-9
Dobaczewski M, Bujak M, Zymek P, Ren G, Entman ML, Frangogiannis NG (2006) Extracellular matrix remodeling in canine and mouse myocardial infarcts. Cell Tissue Res 324:475–488. https://doi.org/10.1007/s00441-005-0144-6
Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG (2010) The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol 48:504–511. https://doi.org/10.1016/j.yjmcc.2009.07.015
Duerrschmid C, Crawford JR, Reineke E, Taffet GE, Trial J, Entman ML, Haudek SB (2013) TNF receptor 1 signaling is critically involved in mediating angiotensin-II-induced cardiac fibrosis. J Mol Cell Cardiol 57:59–67. https://doi.org/10.1016/j.yjmcc.2013.01.006
Dunlay SM, Roger VL, Weston SA, Jiang R, Redfield MM (2012) Longitudinal changes in ejection fraction in heart failure patients with preserved and reduced ejection fraction. Circ Heart Fail 5:720–726. https://doi.org/10.1161/CIRCHEARTFAILURE.111.966366
Dutta P, Nahrendorf M (2015) Monocytes in myocardial infarction. Arterioscler Thromb Vasc Biol 35:1066–1070. https://doi.org/10.1161/ATVBAHA.114.304652
Epelman S, Lavine KJ, Beaudin AE, Sojka DK, Carrero JA, Calderon B, Brija T, Gautier EL, Ivanov S, Satpathy AT, Schilling JD, Schwendener R, Sergin I, Razani B, Forsberg EC, Yokoyama WM, Unanue ER, Colonna M, Randolph GJ, Mann DL (2014) Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40:91–104. https://doi.org/10.1016/j.immuni.2013.11.019
Fabre A, Sheppard MN (2006) Sudden adult death syndrome and other non-ischaemic causes of sudden cardiac death. Heart 92:316–320. https://doi.org/10.1136/hrt.2004.045518
Fairweather D, Rose NR (2007) Coxsackievirus-induced myocarditis in mice: a model of autoimmune disease for studying immunotoxicity. Methods 41:118–122. https://doi.org/10.1016/j.ymeth.2006.07.009
Fan D, Takawale A, Lee J, Kassiri Z (2012) Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenes Tissue Repair 5:15. https://doi.org/10.1186/1755-1536-5-15
Fernandez-Aviles F (2018) Cardiac regeneration in 2017: novel paradigms in the fight against heart failure. Nat Rev Cardiol 15:73–74. https://doi.org/10.1038/nrcardio.2017.218
Ferrari R, Bueno H, Chioncel O, Cleland JG, Stough WG, Lettino M, Metra M, Parissis JT, Pinto F, Ponikowski P, Ruschitzka F, Tavazzi L (2018) Acute heart failure: lessons learned, roads ahead. Eur J Heart Fail 20:842–850. https://doi.org/10.1002/ejhf.1169
Fomovsky GM, Rouillard AD, Holmes JW (2012) Regional mechanics determine collagen fiber structure in healing myocardial infarcts. J Mol Cell Cardiol 52:1083–1090. https://doi.org/10.1016/j.yjmcc.2012.02.012
Frangogiannis NG (2018) Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol Aspects Med. https://doi.org/10.1016/j.mam.2018.07.001
Frangogiannis NG (2018) Cell biological mechanisms in regulation of the post-infarction inflammatory response. Curr Opin Physiol 1:7–13. https://doi.org/10.1016/j.cophys.2017.09.001
Frangogiannis NG (2017) The extracellular matrix in myocardial injury, repair, and remodeling. J Clin Invest 127:1600–1612. https://doi.org/10.1172/JCI87491
Frangogiannis NG (2016) The functional pluralism of fibroblasts in the infarcted myocardium. Circ Res 119:1049–1051. https://doi.org/10.1161/CIRCRESAHA.116.309926
Frangogiannis NG (2008) The immune system and cardiac repair. Pharmacol Res 58:88–111. https://doi.org/10.1016/j.phrs.2008.06.007
Frangogiannis NG (2012) Regulation of the inflammatory response in cardiac repair. Circ Res 110:159–173. https://doi.org/10.1161/CIRCRESAHA.111.243162
Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, Mendoza LH, Spengler RN, Smith CW, Entman ML (1998) Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 98:699–710. https://doi.org/10.1161/01.CIR.98.7.699
Frangogiannis NG, Mendoza LH, Ren G, Akrivakis S, Jackson PL, Michael LH, Smith CW, Entman ML (2003) MCSF expression is induced in healing myocardial infarcts and may regulate monocyte and endothelial cell phenotype. Am J Physiol Heart Circ Physiol 285:H483–H492. https://doi.org/10.1152/ajpheart.01016.2002
Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, Winkelmann K, Michael LH, Lawler J, Entman ML (2005) Critical role of endogenous thrombospondin-1 in preventing expansion of healing myocardial infarcts. Circulation 111:2935–2942. https://doi.org/10.1161/CIRCULATIONAHA.104.510354
Freichel M, Berlin M, Schurger A, Mathar I, Bacmeister L, Medert R, Frede W, Marx A, Segin S, Londono JEC (2017) TRP channels in the heart. In: Emir TLR (ed) Neurobiology of TRP channels. CRC, Boca Rato, pp 149–185
Fung G, Luo H, Qiu Y, Yang D, McManus B (2016) Myocarditis. Circ Res 118:496–514. https://doi.org/10.1161/CIRCRESAHA.115.306573
Fuse K, Chan G, Liu Y, Gudgeon P, Husain M, Chen M, Yeh WC, Akira S, Liu PP (2005) Myeloid differentiation factor-88 plays a crucial role in the pathogenesis of Coxsackievirus B3-induced myocarditis and influences type I interferon production. Circulation 112:2276–2285. https://doi.org/10.1161/CIRCULATIONAHA.105.536433
Gabbiani G (2003) The myofibroblast in wound healing and fibrocontractive diseases. J Pathol 200:500–503. https://doi.org/10.1002/path.1427
Gaggar A, Jackson PL, Noerager BD, O’Reilly PJ, McQuaid DB, Rowe SM, Clancy JP, Blalock JE (2008) A novel proteolytic cascade generates an extracellular matrix-derived chemoattractant in chronic neutrophilic inflammation. J Immunol 180:5662–5669. https://doi.org/10.4049/jimmunol.180.8.5662
Gao XM, Dart AM, Dewar E, Jennings G, Du XJ (2000) Serial echocardiographic assessment of left ventricular dimensions and function after myocardial infarction in mice. Cardiovasc Res 45:330–338. https://doi.org/10.1016/S0008-6363(99)00274-6
Garcia-Menendez L, Karamanlidis G, Kolwicz S, Tian R (2013) Substrain specific response to cardiac pressure overload in C57BL/6 mice. Am J Physiol Heart Circ Physiol 305:H397–H402. https://doi.org/10.1152/ajpheart.00088.2013
Garlie JB, Hamid T, Gu Y, Ismahil MA, Chandrasekar B, Prabhu SD (2011) Tumor necrosis factor receptor 2 signaling limits beta-adrenergic receptor-mediated cardiac hypertrophy in vivo. Basic Res Cardiol 106:1193–1205. https://doi.org/10.1007/s00395-011-0196-6
Garmaroudi FS, Marchant D, Hendry R, Luo H, Yang D, Ye X, Shi J, McManus BM (2015) Coxsackievirus B3 replication and pathogenesis. Future Microbiol 10:629–653. https://doi.org/10.2217/fmb.15.5
Geerling JC, Engeland WC, Kawata M, Loewy AD (2006) Aldosterone target neurons in the nucleus tractus solitarius drive sodium appetite. J Neurosci 26:411–417. https://doi.org/10.1523/JNEUROSCI.3115-05.2006
Gehrmann J, Frantz S, Maguire CT, Vargas M, Ducharme A, Wakimoto H, Lee RT, Berul CI (2001) Electrophysiological characterization of murine myocardial ischemia and infarction. Basic Res Cardiol 96:237–250. https://doi.org/10.1007/s003950170054
Gharacholou SM, Alexander KP, Chen AY, Wang TY, Melloni C, Gibler WB, Pollack CV Jr, Ohman EM, Peterson ED, Roe MT (2010) Implications and reasons for the lack of use of reperfusion therapy in patients with ST-segment elevation myocardial infarction: findings from the CRUSADE initiative. Am Heart J 159:757–763. https://doi.org/10.1016/j.ahj.2010.02.009
Gill SE, Parks WC (2008) Metalloproteinases and their inhibitors: regulators of wound healing. Int J Biochem Cell Biol 40:1334–1347. https://doi.org/10.1016/j.biocel.2007.10.024
Gomolak JR, Didion SP (2014) Angiotensin II-induced endothelial dysfunction is temporally linked with increases in interleukin-6 and vascular macrophage accumulation. Front Physiol 5:396. https://doi.org/10.3389/fphys.2014.00396
Gonzalez GE, Rhaleb NE, D’Ambrosio MA, Nakagawa P, Liu Y, Leung P, Dai X, Yang XP, Peterson EL, Carretero OA (2015) Deletion of interleukin-6 prevents cardiac inflammation, fibrosis and dysfunction without affecting blood pressure in angiotensin II-high salt-induced hypertension. J Hypertens 33:144–152. https://doi.org/10.1097/HJH.0000000000000358
Gordon JW, Shaw JA, Kirshenbaum LA (2011) Multiple facets of NF-kappaB in the heart: to be or not to NF-kappaB. Circ Res 108:1122–1132. https://doi.org/10.1161/CIRCRESAHA.110.226928
Grisanti LA, Gumpert AM, Traynham CJ, Gorsky JE, Repas AA, Gao E, Carter RL, Yu D, Calvert JW, Garcia AP, Ibanez B, Rabinowitz JE, Koch WJ, Tilley DG (2016) Leukocyte-expressed beta2-adrenergic receptors are essential for survival after acute myocardial injury. Circulation 134:153–167. https://doi.org/10.1161/CIRCULATIONAHA.116.022304
Grisanti LA, Traynham CJ, Repas AA, Gao E, Koch WJ, Tilley DG (2016) beta2-Adrenergic receptor-dependent chemokine receptor 2 expression regulates leukocyte recruitment to the heart following acute injury. Proc Natl Acad Sci USA 113:15126–15131. https://doi.org/10.1073/pnas.1611023114
Gurantz D, Cowling RT, Varki N, Frikovsky E, Moore CD, Greenberg BH (2005) IL-1beta and TNF-alpha upregulate angiotensin II type 1 (AT1) receptors on cardiac fibroblasts and are associated with increased AT1 density in the post-MI heart. J Mol Cell Cardiol 38:505–515. https://doi.org/10.1016/j.yjmcc.2004.12.015
Guyenet PG (2006) The sympathetic control of blood pressure. Nat Rev Neurosci 7:335–346. https://doi.org/10.1038/nrn1902
Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG (2007) Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med 204:2449–2460. https://doi.org/10.1084/jem.20070657
Hanif W, Alex L, Su Y, Shinde AV, Russo I, Li N, Frangogiannis NG (2017) Left atrial remodeling, hypertrophy, and fibrosis in mouse models of heart failure. Cardiovasc Pathol 30:27–37. https://doi.org/10.1016/j.carpath.2017.06.003
Haudek SB, Cheng J, Du J, Wang Y, Hermosillo-Rodriguez J, Trial J, Taffet GE, Entman ML (2010) Monocytic fibroblast precursors mediate fibrosis in angiotensin-II-induced cardiac hypertrophy. J Mol Cell Cardiol 49:499–507. https://doi.org/10.1016/j.yjmcc.2010.05.005
Hausenloy DJ, Yellon DM (2013) Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest 123:92–100. https://doi.org/10.1172/JCI62874
He L, Kim T, Long Q, Liu J, Wang P, Zhou Y, Ding Y, Prasain J, Wood PA, Yang Q (2012) Carnitine palmitoyltransferase-1b deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity. Circulation 126:1705–1716. https://doi.org/10.1161/CIRCULATIONAHA.111.075978
Hermans H, Swinnen M, Pokreisz P, Caluwe E, Dymarkowski S, Herregods MC, Janssens S (1985) Herijgers P (2014) Murine pressure overload models: a 30-MHz look brings a whole new “sound” into data interpretation. J Appl Physiol 117:563–571. https://doi.org/10.1152/japplphysiol.00363.2014
Hermida N, Michel L, Esfahani H, Dubois-Deruy E, Hammond J, Bouzin C, Markl A, Colin H, Steenbergen AV, De Meester C, Beauloye C, Horman S, Yin X, Mayr M, Balligand JL (2018) Cardiac myocyte beta3-adrenergic receptors prevent myocardial fibrosis by modulating oxidant stress-dependent paracrine signaling. Eur Heart J 39:888–898. https://doi.org/10.1093/eurheartj/ehx366
Heusch G, Gersh BJ (2017) The pathophysiology of acute myocardial infarction and strategies of protection beyond reperfusion: a continual challenge. Eur Heart J 38:774–784. https://doi.org/10.1093/eurheartj/ehw224
Heusch G, Libby P, Gersh B, Yellon D, Bohm M, Lopaschuk G, Opie L (2014) Cardiovascular remodelling in coronary artery disease and heart failure. Lancet 383:1933–1943. https://doi.org/10.1016/S0140-6736(14)60107-0
Hinrichs S, Scherschel K, Kruger S, Neumann JT, Schwarzl M, Yan I, Warnke S, Ojeda FM, Zeller T, Karakas M, Keller T, Meyer C, Blankenberg S, Westermann D, Lindner D (2018) Precursor proadrenomedullin influences cardiomyocyte survival and local inflammation related to myocardial infarction. Proc Natl Acad Sci USA 115:E8727–E8736. https://doi.org/10.1073/pnas.1721635115
Hinz B (2010) The myofibroblast: paradigm for a mechanically active cell. J Biomech 43:146–155. https://doi.org/10.1016/j.jbiomech.2009.09.020
Hinz B, Mastrangelo D, Iselin CE, Chaponnier C, Gabbiani G (2001) Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am J Pathol 159:1009–1020. https://doi.org/10.1016/S0002-9440(10)61776-2
Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, Gabbiani G (2007) The myofibroblast: one function, multiple origins. Am J Pathol 170:1807–1816. https://doi.org/10.2353/ajpath.2007.070112
Ho JE, Enserro D, Brouwers FP, Kizer JR, Shah SJ, Psaty BM, Bartz TM, Santhanakrishnan R, Lee DS, Chan C, Liu K, Blaha MJ, Hillege HL, van der Harst P, van Gilst WH, Kop WJ, Gansevoort RT, Vasan RS, Gardin JM, Levy D, Gottdiener JS, de Boer RA, Larson MG (2016) Predicting Heart Failure With Preserved and Reduced Ejection Fraction: The International Collaboration on Heart Failure Subtypes. Circ Heart Fail. https://doi.org/10.1161/circheartfailure.115.003116
Hofmann U, Frantz S (2015) Role of lymphocytes in myocardial injury, healing, and remodeling after myocardial infarction. Circ Res 116:354–367. https://doi.org/10.1161/CIRCRESAHA.116.304072
Horckmans M, Ring L, Duchene J, Santovito D, Schloss MJ, Drechsler M, Weber C, Soehnlein O, Steffens S (2017) Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur Heart J 38:187–197. https://doi.org/10.1093/eurheartj/ehw002
Houde M, Schwertani A, Touil H, Desbiens L, Sarrhini O, Lecomte R, Lepage M, Gagnon H, Takai S, Pejler G, Jacques D, Gobeil F Jr, Day R, D’Orleans-Juste P (2018) Mouse mast cell protease 4 deletion protects heart function and survival after permanent myocardial infarction. Front Pharmacol 9:868. https://doi.org/10.3389/fphar.2018.00868
Howangyin KY, Zlatanova I, Pinto C, Ngkelo A, Cochain C, Rouanet M, Vilar J, Lemitre M, Stockmann C, Fleischmann BK, Mallat Z, Silvestre JS (2016) Myeloid-epithelial-reproductive receptor tyrosine kinase and milk fat globule epidermal growth factor 8 coordinately improve remodeling after myocardial infarction via local delivery of vascular endothelial growth factor. Circulation 133:826–839. https://doi.org/10.1161/CIRCULATIONAHA.115.020857
Huang CH, Vallejo JG, Kollias G, Mann DL (2009) Role of the innate immune system in acute viral myocarditis. Basic Res Cardiol 104:228–237. https://doi.org/10.1007/s00395-008-0765-5
Huang Y, Wright CD, Merkwan CL, Baye NL, Liang Q, Simpson PC, O’Connell TD (2007) An alpha1A-adrenergic-extracellular signal-regulated kinase survival signaling pathway in cardiac myocytes. Circulation 115:763–772. https://doi.org/10.1161/CIRCULATIONAHA.106.664862
Huber SA, Sartini D (2005) Roles of tumor necrosis factor alpha (TNF-alpha) and the p55 TNF receptor in CD1d induction and coxsackievirus B3-induced myocarditis. J Virol 79:2659–2665. https://doi.org/10.1128/JVI.79.5.2659-2665.2005
Huebener P, Abou-Khamis T, Zymek P, Bujak M, Ying X, Chatila K, Haudek S, Thakker G, Frangogiannis NG (2008) CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. J Immunol 180:2625–2633. https://doi.org/10.4049/jimmunol.180.4.2625
Hufnagel G, Chapman N, Tracy S (1995) A non-cardiovirulent strain of coxsackievirus B3 causes myocarditis in mice with severe combined immunodeficiency syndrome. Eur Heart J 16(Suppl O):18–19. https://doi.org/10.1093/eurheartj/16.suppl_o.18
Huynh ML, Fadok VA, Henson PM (2002) Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest 109:41–50. https://doi.org/10.1172/JCI11638
Ibanez B, Heusch G, Ovize M, Van de Werf F (2015) Evolving therapies for myocardial ischemia/reperfusion injury. J Am Coll Cardiol 65:1454–1471. https://doi.org/10.1016/j.jacc.2015.02.032
Ibanez B, James S, Agewall S, Antunes MJ, Bucciarelli-Ducci C, Bueno H, Caforio ALP, Crea F, Goudevenos JA, Halvorsen S, Hindricks G, Kastrati A, Lenzen MJ, Prescott E, Roffi M, Valgimigli M, Varenhorst C, Vranckx P, Widimsky P, Group ESCSD (2018) 2017 ESC Guidelines for the management of acute myocardial infarction in patients presenting with ST-segment elevation: The Task Force for the management of acute myocardial infarction in patients presenting with ST-segment elevation of the European Society of Cardiology (ESC). Eur Heart J 39:119–177. https://doi.org/10.1093/eurheartj/ehx393
Ikeuchi M, Tsutsui H, Shiomi T, Matsusaka H, Matsushima S, Wen J, Kubota T, Takeshita A (2004) Inhibition of TGF-beta signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc Res 64:526–535. https://doi.org/10.1016/j.cardiores.2004.07.017
Imai Y, Kariya T, Iwakiri M, Yamada Y, Takimoto E (2018) Sildenafil ameliorates right ventricular early molecular derangement during left ventricular pressure overload. PLoS One 13:e0195528. https://doi.org/10.1371/journal.pone.0195528
Iwanaga Y, Aoyama T, Kihara Y, Onozawa Y, Yoneda T, Sasayama S (2002) Excessive activation of matrix metalloproteinases coincides with left ventricular remodeling during transition from hypertrophy to heart failure in hypertensive rats. J Am Coll Cardiol 39:1384–1391. https://doi.org/10.1016/S0735-1097(02)01756-4
Iyer RP, de Castro Bras LE, Cannon PL, Ma Y, DeLeon-Pennell KY, Jung M, Flynn ER, Henry JB, Bratton DR, White JA, Fulton LK, Grady AW, Lindsey ML (2016) Defining the sham environment for post-myocardial infarction studies in mice. Am J Physiol Heart Circ Physiol 311:H822–H836. https://doi.org/10.1152/ajpheart.00067.2016
Jakel S, Kuckelkorn U, Szalay G, Plotz M, Textoris-Taube K, Opitz E, Klingel K, Stevanovic S, Kandolf R, Kotsch K, Stangl K, Kloetzel PM, Voigt A (2009) Differential interferon responses enhance viral epitope generation by myocardial immunoproteasomes in murine enterovirus myocarditis. Am J Pathol 175:510–518. https://doi.org/10.2353/ajpath.2009.090033
Janicki JS, Brower GL (2002) The role of myocardial fibrillar collagen in ventricular remodeling and function. J Card Fail 8:S319–S325. https://doi.org/10.1054/jcaf.2002.129260
Jaquenod De Giusti C, Ure AE, Rivadeneyra L, Schattner M, Gomez RM (2015) Macrophages and galectin 3 play critical roles in CVB3-induced murine acute myocarditis and chronic fibrosis. J Mol Cell Cardiol 85:58–70. https://doi.org/10.1016/j.yjmcc.2015.05.010
Jeong EM, Monasky MM, Gu L, Taglieri DM, Patel BG, Liu H, Wang Q, Greener I, Dudley SC Jr, Solaro RJ (2013) Tetrahydrobiopterin improves diastolic dysfunction by reversing changes in myofilament properties. J Mol Cell Cardiol 56:44–54. https://doi.org/10.1016/j.yjmcc.2012.12.003
Joiner ML, Koval OM, Li J, He BJ, Allamargot C, Gao Z, Luczak ED, Hall DD, Fink BD, Chen B, Yang J, Moore SA, Scholz TD, Strack S, Mohler PJ, Sivitz WI, Song LS, Anderson ME (2012) CaMKII determines mitochondrial stress responses in heart. Nature 491:269–273. https://doi.org/10.1038/nature11444
Jong WM, Ten Cate H, Linnenbank AC, de Boer OJ, Reitsma PH, de Winter RJ, Zuurbier CJ (2016) Reduced acute myocardial ischemia-reperfusion injury in IL-6-deficient mice employing a closed-chest model. Inflamm Res 65:489–499. https://doi.org/10.1007/s00011-016-0931-4
Jugdutt BI (2010) Preventing adverse remodeling and rupture during healing after myocardial infarction in mice and humans. Circulation 122:103–105. https://doi.org/10.1161/CIRCULATIONAHA.110.969410
Kallikourdis M, Martini E, Carullo P, Sardi C, Roselli G, Greco CM, Vignali D, Riva F, Ormbostad Berre AM, Stolen TO, Fumero A, Faggian G, Di Pasquale E, Elia L, Rumio C, Catalucci D, Papait R, Condorelli G (2017) T cell costimulation blockade blunts pressure overload-induced heart failure. Nat Commun 8:14680. https://doi.org/10.1038/ncomms14680
Kandolf R, Hofschneider PH (1985) Molecular cloning of the genome of a cardiotropic Coxsackie B3 virus: full-length reverse-transcribed recombinant cDNA generates infectious virus in mammalian cells. Proc Natl Acad Sci USA 82:4818–4822. https://doi.org/10.1073/pnas.82.14.4818
Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, Uematsu S, Jung A, Kawai T, Ishii KJ, Yamaguchi O, Otsu K, Tsujimura T, Koh CS, Reis e Sousa C, Matsuura Y, Fujita T, Akira S (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441:101–105. https://doi.org/10.1038/nature04734
Kazakov A, Hall RA, Werner C, Meier T, Trouvain A, Rodionycheva S, Nickel A, Lammert F, Maack C, Bohm M, Laufs U (2018) Raf kinase inhibitor protein mediates myocardial fibrosis under conditions of enhanced myocardial oxidative stress. Basic Res Cardiol 113:42. https://doi.org/10.1007/s00395-018-0700-3
Kemp CD, Conte JV (2012) The pathophysiology of heart failure. Cardiovasc Pathol 21:365–371. https://doi.org/10.1016/j.carpath.2011.11.007
Kinet JP (2007) The essential role of mast cells in orchestrating inflammation. Immunol Rev 217:5–7. https://doi.org/10.1111/j.1600-065X.2007.00528.x
Kishimoto C, Kuribayashi K, Masuda T, Tomioka N, Kawai C (1985) Immunologic behavior of lymphocytes in experimental viral myocarditis: significance of T lymphocytes in the severity of myocarditis and silent myocarditis in BALB/c-nu/nu mice. Circulation 71:1247–1254. https://doi.org/10.1161/01.CIR.71.6.1247
Klingel K, Hohenadl C, Canu A, Albrecht M, Seemann M, Mall G, Kandolf R (1992) Ongoing enterovirus-induced myocarditis is associated with persistent heart muscle infection: quantitative analysis of virus replication, tissue damage, and inflammation. Proc Natl Acad Sci USA 89:314–318. https://doi.org/10.1073/pnas.89.1.314
Klocke R, Tian W, Kuhlmann MT, Nikol S (2007) Surgical animal models of heart failure related to coronary heart disease. Cardiovasc Res 74:29–38. https://doi.org/10.1016/j.cardiores.2006.11.026
Knight WE, Chen S, Zhang Y, Oikawa M, Wu M, Zhou Q, Miller CL, Cai Y, Mickelsen DM, Moravec C, Small EM, Abe J, Yan C (2016) PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc Natl Acad Sci USA 113:E7116–E7125. https://doi.org/10.1073/pnas.1607728113
Koitabashi N, Danner T, Zaiman AL, Pinto YM, Rowell J, Mankowski J, Zhang D, Nakamura T, Takimoto E, Kass DA (2011) Pivotal role of cardiomyocyte TGF-beta signaling in the murine pathological response to sustained pressure overload. J Clin Invest 121:2301–2312. https://doi.org/10.1172/JCI44824
Kong P, Christia P, Frangogiannis NG (2014) The pathogenesis of cardiac fibrosis. Cell Mol Life Sci 71:549–574. https://doi.org/10.1007/s00018-013-1349-6
Kong P, Shinde AV, Su Y, Russo I, Chen B, Saxena A, Conway SJ, Graff JM, Frangogiannis NG (2018) Opposing actions of fibroblast and cardiomyocyte Smad3 signaling in the infarcted myocardium. Circulation 137:707–724. https://doi.org/10.1161/CIRCULATIONAHA.117.029622
Kraft L, Erdenesukh T, Sauter M, Tschope C, Klingel K (2019) Blocking the IL-1beta signalling pathway prevents chronic viral myocarditis and cardiac remodeling. Basic Res Cardiol 114:11. https://doi.org/10.1007/s00395-019-0719-0
Krysko DV, Agostinis P, Krysko O, Garg AD, Bachert C, Lambrecht BN, Vandenabeele P (2011) Emerging role of damage-associated molecular patterns derived from mitochondria in inflammation. Trends Immunol 32:157–164. https://doi.org/10.1016/j.it.2011.01.005
Kubota T, Bounoutas GS, Miyagishima M, Kadokami T, Sanders VJ, Bruton C, Robbins PD, McTiernan CF, Feldman AM (2000) Soluble tumor necrosis factor receptor abrogates myocardial inflammation but not hypertrophy in cytokine-induced cardiomyopathy. Circulation 101:2518–2525. https://doi.org/10.1161/01.CIR.101.21.2518
Kumar AG, Ballantyne CM, Michael LH, Kukielka GL, Youker KA, Lindsey ML, Hawkins HK, Birdsall HH, MacKay CR, LaRosa GJ, Rossen RD, Smith CW, Entman ML (1997) Induction of monocyte chemoattractant protein-1 in the small veins of the ischemic and reperfused canine myocardium. Circulation 95:693–700. https://doi.org/10.1161/01.CIR.95.3.693
Kurisu S, Ozono R, Oshima T, Kambe M, Ishida T, Sugino H, Matsuura H, Chayama K, Teranishi Y, Iba O, Amano K, Matsubara H (2003) Cardiac angiotensin II type 2 receptor activates the kinin/NO system and inhibits fibrosis. Hypertension 41:99–107. https://doi.org/10.1161/01.HYP.0000050101.90932.14
Kzhyshkowska J, Workman G, Cardo-Vila M, Arap W, Pasqualini R, Gratchev A, Krusell L, Goerdt S, Sage EH (2006) Novel function of alternatively activated macrophages: stabilin-1-mediated clearance of SPARC. J Immunol 176:5825–5832. https://doi.org/10.4049/jimmunol.176.10.5825
Lang C, Sauter M, Szalay G, Racchi G, Grassi G, Rainaldi G, Mercatanti A, Lang F, Kandolf R, Klingel K (2008) Connective tissue growth factor: a crucial cytokine-mediating cardiac fibrosis in ongoing enterovirus myocarditis. J Mol Med (Berl) 86:49–60. https://doi.org/10.1007/s00109-007-0249-3
Laroumanie F, Douin-Echinard V, Pozzo J, Lairez O, Tortosa F, Vinel C, Delage C, Calise D, Dutaur M, Parini A, Pizzinat N (2014) CD4 + T cells promote the transition from hypertrophy to heart failure during chronic pressure overload. Circulation 129:2111–2124. https://doi.org/10.1161/CIRCULATIONAHA.113.007101
Leask A (2015) Getting to the heart of the matter: new insights into cardiac fibrosis. Circ Res 116:1269–1276. https://doi.org/10.1161/CIRCRESAHA.116.305381
Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, Lee KM, Kim JI, Markmann JF, Marinelli B, Panizzi P, Lee WW, Iwamoto Y, Milstein S, Epstein-Barash H, Cantley W, Wong J, Cortez-Retamozo V, Newton A, Love K, Libby P, Pittet MJ, Swirski FK, Koteliansky V, Langer R, Weissleder R, Anderson DG, Nahrendorf M (2011) Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol 29:1005–1010. https://doi.org/10.1038/nbt.1989
Levick SP, Melendez GC, Plante E, McLarty JL, Brower GL, Janicki JS (2011) Cardiac mast cells: the centrepiece in adverse myocardial remodelling. Cardiovasc Res 89:12–19. https://doi.org/10.1093/cvr/cvq272
Li J, Brown LF, Hibberd MG, Grossman JD, Morgan JP, Simons M (1996) VEGF, flk-1, and flt-1 expression in a rat myocardial infarction model of angiogenesis. Am J Physiol 270:H1803–H1811. https://doi.org/10.1152/ajpheart.1996.270.5.H1803
Li L, Guo X, Chen Y, Yin H, Li J, Doan J, Liu Q (2016) Assessment of cardiac morphological and functional changes in mouse model of transverse aortic constriction by echocardiographic imaging. J Vis Exp. https://doi.org/10.3791/54101
Liao Y, Takashima S, Maeda N, Ouchi N, Komamura K, Shimomura I, Hori M, Matsuzawa Y, Funahashi T, Kitakaze M (2005) Exacerbation of heart failure in adiponectin-deficient mice due to impaired regulation of AMPK and glucose metabolism. Cardiovasc Res 67:705–713. https://doi.org/10.1016/j.cardiores.2005.04.018
Lim BK, Choe SC, Shin JO, Ho SH, Kim JM, Yu SS, Kim S, Jeon ES (2002) Local expression of interleukin-1 receptor antagonist by plasmid DNA improves mortality and decreases myocardial inflammation in experimental coxsackieviral myocarditis. Circulation 105:1278–1281. https://doi.org/10.1161/circ.105.11.1278
Lindner D, Hilbrandt M, Marggraf K, Becher PM, Hilfiker-Kleiner D, Klingel K, Pauschinger M, Schultheiss HP, Tschope C, Westermann D (2012) Protective function of STAT3 in CVB3-induced myocarditis. Cardiol Res Pract 2012:437623. https://doi.org/10.1155/2012/437623
Lindner D, Li J, Savvatis K, Klingel K, Blankenberg S, Tschope C, Westermann D (2014) Cardiac fibroblasts aggravate viral myocarditis: cell specific coxsackievirus B3 replication. Mediat Inflamm 2014:519528. https://doi.org/10.1155/2014/519528
Lindner D, Westermann D (2015) Firefighting in viral myocarditis—extinguish galectin-3 to control the inflamed heart? J Mol Cell Cardiol 85:226–228. https://doi.org/10.1016/j.yjmcc.2015.06.004
Lindner D, Zietsch C, Tank J, Sossalla S, Fluschnik N, Hinrichs S, Maier L, Poller W, Blankenberg S, Schultheiss HP, Tschope C, Westermann D (2014) Cardiac fibroblasts support cardiac inflammation in heart failure. Basic Res Cardiol 109:428. https://doi.org/10.1007/s00395-014-0428-7
Lindsey ML (2018) Assigning matrix metalloproteinase roles in ischaemic cardiac remodelling. Nat Rev Cardiol 15:471–479. https://doi.org/10.1038/s41569-018-0022-z
Lindsey ML, Bolli R, Canty JM Jr, Du XJ, Frangogiannis NG, Frantz S, Gourdie RG, Holmes JW, Jones SP, Kloner RA, Lefer DJ, Liao R, Murphy E, Ping P, Przyklenk K, Recchia FA, Schwartz Longacre L, Ripplinger CM, Van Eyk JE, Heusch G (2018) Guidelines for experimental models of myocardial ischemia and infarction. Am J Physiol Heart Circ Physiol 314:H812–H838. https://doi.org/10.1152/ajpheart.00335.2017
Lindsey ML, Jung M, Yabluchanskiy A, Cannon PL, Iyer RP, Flynn ER, DeLeon-Pennell KY, Valerio FM, Harrison CL, Ripplinger CM, Hall ME, Ma Y (2019) Exogenous CXCL4 infusion inhibits macrophage phagocytosis by limiting CD36 signalling to enhance post-myocardial infarction cardiac dilation and mortality. Cardiovasc Res 115:395–408. https://doi.org/10.1093/cvr/cvy211
Lindsey ML, Kassiri Z, Virag JAI, de Castro Bras LE, Scherrer-Crosbie M (2018) Guidelines for measuring cardiac physiology in mice. Am J Physiol Heart Circ Physiol 314:H733–H752. https://doi.org/10.1152/ajpheart.00339.2017
Lipps C, Nguyen JH, Pyttel L, Lynch TLT, Liebetrau C, Aleshcheva G, Voss S, Dorr O, Nef HM, Mollmann H, Hamm CW, Sadayappan S, Troidl C (2016) N-terminal fragment of cardiac myosin binding protein-C triggers pro-inflammatory responses in vitro. J Mol Cell Cardiol 99:47–56. https://doi.org/10.1016/j.yjmcc.2016.09.003
Lopez B, Querejeta R, Gonzalez A, Larman M, Diez J (2012) Collagen cross-linking but not collagen amount associates with elevated filling pressures in hypertensive patients with stage C heart failure: potential role of lysyl oxidase. Hypertension 60:677–683. https://doi.org/10.1161/HYPERTENSIONAHA.112.196113
Lother A, Berger S, Gilsbach R, Rosner S, Ecke A, Barreto F, Bauersachs J, Schutz G, Hein L (2011) Ablation of mineralocorticoid receptors in myocytes but not in fibroblasts preserves cardiac function. Hypertension 57:746–754. https://doi.org/10.1161/HYPERTENSIONAHA.110.163287
Lother A, Furst D, Bergemann S, Gilsbach R, Grahammer F, Huber TB, Hilgendorf I, Bode C, Moser M, Hein L (2016) Deoxycorticosterone acetate/salt-induced cardiac but not renal injury is mediated by endothelial mineralocorticoid receptors independently from blood pressure. Hypertension 67:130–138. https://doi.org/10.1161/HYPERTENSIONAHA.115.06530
Lovelock JD, Monasky MM, Jeong EM, Lardin HA, Liu H, Patel BG, Taglieri DM, Gu L, Kumar P, Pokhrel N, Zeng D, Belardinelli L, Sorescu D, Solaro RJ, Dudley SC Jr (2012) Ranolazine improves cardiac diastolic dysfunction through modulation of myofilament calcium sensitivity. Circ Res 110:841–850. https://doi.org/10.1161/CIRCRESAHA.111.258251
Lutgens E, Daemen MJ, de Muinck ED, Debets J, Leenders P, Smits JF (1999) Chronic myocardial infarction in the mouse: cardiac structural and functional changes. Cardiovasc Res 41:586–593. https://doi.org/10.1016/S0008-6363(98)00216-8
Lymperopoulos A, Rengo G, Koch WJ (2013) Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res 113:739–753. https://doi.org/10.1161/CIRCRESAHA.113.300308
Lyons RM, Keski-Oja J, Moses HL (1988) Proteolytic activation of latent transforming growth factor-beta from fibroblast-conditioned medium. J Cell Biol 106:1659–1665. https://doi.org/10.1083/jcb.106.5.1659
Ma J, Luo T, Zeng Z, Fu H, Asano Y, Liao Y, Minamino T, Kitakaze M (2016) Histone deacetylase inhibitor phenylbutyrate exaggerates heart failure in pressure overloaded mice independently of HDAC inhibition. Sci Rep 6:34036. https://doi.org/10.1038/srep34036
Ma Y, Yabluchanskiy A, Iyer RP, Cannon PL, Flynn ER, Jung M, Henry J, Cates CA, Deleon-Pennell KY, Lindsey ML (2016) Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res 110:51–61. https://doi.org/10.1093/cvr/cvw024
Mann DL (2011) The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls. Circ Res 108:1133–1145. https://doi.org/10.1161/CIRCRESAHA.110.226936
Mann DL, Bristow MR (2005) Mechanisms and models in heart failure: the biomechanical model and beyond. Circulation 111:2837–2849. https://doi.org/10.1161/CIRCULATIONAHA.104.500546
Marchant DJ, Boyd JH, Lin DC, Granville DJ, Garmaroudi FS, McManus BM (2012) Inflammation in myocardial diseases. Circ Res 110:126–144. https://doi.org/10.1161/CIRCRESAHA.111.243170
Martino MM, Briquez PS, Ranga A, Lutolf MP, Hubbell JA (2013) Heparin-binding domain of fibrin(ogen) binds growth factors and promotes tissue repair when incorporated within a synthetic matrix. Proc Natl Acad Sci USA 110:4563–4568. https://doi.org/10.1073/pnas.1221602110
Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ, Harrison DG (2010) Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ Res 107:263–270. https://doi.org/10.1161/CIRCRESAHA.110.217299
McEwan PE, Gray GA, Sherry L, Webb DJ, Kenyon CJ (1998) Differential effects of angiotensin II on cardiac cell proliferation and intramyocardial perivascular fibrosis in vivo. Circulation 98:2765–2773. https://doi.org/10.1161/01.CIR.98.24.2765
Mehrad B, Keane MP, Strieter RM (2007) Chemokines as mediators of angiogenesis. Thromb Haemost 97:755–762. https://doi.org/10.1160/TH07-01-0040
Melleby AO, Romaine A, Aronsen JM, Veras I, Zhang L, Sjaastad I, Lunde IG, Christensen G (2018) A novel method for high precision aortic constriction that allows for generation of specific cardiac phenotypes in mice. Cardiovasc Res 114:1680–1690. https://doi.org/10.1093/cvr/cvy141
Melnick JL (1983) Portraits of viruses: the picornaviruses. Intervirology 20:61–100. https://doi.org/10.1159/000149376
Meng X, Yang J, Dong M, Zhang K, Tu E, Gao Q, Chen W, Zhang C, Zhang Y (2016) Regulatory T cells in cardiovascular diseases. Nat Rev Cardiol 13:167–179. https://doi.org/10.1038/nrcardio.2015.169
Mersmann J, Habeck K, Latsch K, Zimmermann R, Jacoby C, Fischer JW, Hartmann C, Schrader J, Kirschning CJ, Zacharowski K (2011) Left ventricular dilation in toll-like receptor 2 deficient mice after myocardial ischemia/reperfusion through defective scar formation. Basic Res Cardiol 106:89–98. https://doi.org/10.1007/s00395-010-0127-y
Michael LH, Entman ML, Hartley CJ, Youker KA, Zhu J, Hall SR, Hawkins HK, Berens K, Ballantyne CM (1995) Myocardial ischemia and reperfusion: a murine model. Am J Physiol 269:H2147–H2154. https://doi.org/10.1152/ajpheart.1995.269.6.H2147
Mohammed SF, Ohtani T, Korinek J, Lam CS, Larsen K, Simari RD, Valencik ML, Burnett JC Jr, Redfield MM (2010) Mineralocorticoid accelerates transition to heart failure with preserved ejection fraction via “nongenomic effects”. Circulation 122:370–378. https://doi.org/10.1161/CIRCULATIONAHA.109.915215
Mohammed SF, Storlie JR, Oehler EA, Bowen LA, Korinek J, Lam CS, Simari RD, Burnett JC Jr, Redfield MM (2012) Variable phenotype in murine transverse aortic constriction. Cardiovasc Pathol 21:188–198. https://doi.org/10.1016/j.carpath.2011.05.002
Mollenhauer M, Friedrichs K, Lange M, Gesenberg J, Remane L, Kerkenpass C, Krause J, Schneider J, Ravekes T, Maass M, Halbach M, Peinkofer G, Saric T, Mehrkens D, Adam M, Deuschl FG, Lau D, Geertz B, Manchanda K, Eschenhagen T, Kubala L, Rudolph TK, Wu Y, Tang WHW, Hazen SL, Baldus S, Klinke A, Rudolph V (2017) Myeloperoxidase mediates postischemic arrhythmogenic ventricular remodeling. Circ Res 121:56–70. https://doi.org/10.1161/CIRCRESAHA.117.310870
Mollmann H, Nef HM, Kostin S, von Kalle C, Pilz I, Weber M, Schaper J, Hamm CW, Elsasser A (2006) Bone marrow-derived cells contribute to infarct remodelling. Cardiovasc Res 71:661–671. https://doi.org/10.1016/j.cardiores.2006.06.013
Montecucco F, Carbone F, Schindler TH (2016) Pathophysiology of ST-segment elevation myocardial infarction: novel mechanisms and treatments. Eur Heart J 37:1268–1283. https://doi.org/10.1093/eurheartj/ehv592
Moore-Morris T, Guimaraes-Camboa N, Banerjee I, Zambon AC, Kisseleva T, Velayoudon A, Stallcup WB, Gu Y, Dalton ND, Cedenilla M, Gomez-Amaro R, Zhou B, Brenner DA, Peterson KL, Chen J, Evans SM (2014) Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J Clin Invest 124:2921–2934. https://doi.org/10.1172/JCI74783
Moore JBT, Tang XL, Zhao J, Fischer AG, Wu WJ, Uchida S, Gumpert AM, Stowers H, Wysoczynski M, Bolli R (2018) Epigenetically modified cardiac mesenchymal stromal cells limit myocardial fibrosis and promote functional recovery in a model of chronic ischemic cardiomyopathy. Basic Res Cardiol 114:3. https://doi.org/10.1007/s00395-018-0710-1
Mouton AJ, DeLeon-Pennell KY, Rivera Gonzalez OJ, Flynn ER, Freeman TC, Saucerman JJ, Garrett MR, Ma Y, Harmancey R, Lindsey ML (2018) Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res Cardiol 113:26. https://doi.org/10.1007/s00395-018-0686-x
Muller J, Gorressen S, Grandoch M, Feldmann K, Kretschmer I, Lehr S, Ding Z, Schmitt JP, Schrader J, Garbers C, Heusch G, Kelm M, Scheller J, Fischer JW (2014) Interleukin-6-dependent phenotypic modulation of cardiac fibroblasts after acute myocardial infarction. Basic Res Cardiol 109:440. https://doi.org/10.1007/s00395-014-0440-y
Murdoch CE, Chaubey S, Zeng L, Yu B, Ivetic A, Walker SJ, Vanhoutte D, Heymans S, Grieve DJ, Cave AC, Brewer AC, Zhang M, Shah AM (2014) Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J Am Coll Cardiol 63:2734–2741. https://doi.org/10.1016/j.jacc.2014.02.572
Murphy AM, Wong AL, Bezuhly M (2015) Modulation of angiotensin II signaling in the prevention of fibrosis. Fibrogenes Tissue Repair 8:7. https://doi.org/10.1186/s13069-015-0023-z
Murray DR, Prabhu SD, Chandrasekar B (2000) Chronic beta-adrenergic stimulation induces myocardial proinflammatory cytokine expression. Circulation 101:2338–2341. https://doi.org/10.1161/01.CIR.101.20.2338
Nagatomo Y, Carabello BA, Coker ML, McDermott PJ, Nemoto S, Hamawaki M, Spinale FG (2000) Differential effects of pressure or volume overload on myocardial MMP levels and inhibitory control. Am J Physiol Heart Circ Physiol 278:H151–H161. https://doi.org/10.1152/ajpheart.2000.278.1.H151
Nakaya M, Watari K, Tajima M, Nakaya T, Matsuda S, Ohara H, Nishihara H, Yamaguchi H, Hashimoto A, Nishida M, Nagasaka A, Horii Y, Ono H, Iribe G, Inoue R, Tsuda M, Inoue K, Tanaka A, Kuroda M, Nagata S, Kurose H (2017) Cardiac myofibroblast engulfment of dead cells facilitates recovery after myocardial infarction. J Clin Invest 127:383–401. https://doi.org/10.1172/JCI83822
Nathan C, Ding A (2010) Nonresolving inflammation. Cell 140:871–882. https://doi.org/10.1016/j.cell.2010.02.029
Neumann FJ, Sousa-Uva M, Ahlsson A, Alfonso F, Banning AP, Benedetto U, Byrne RA, Collet JP, Falk V, Head SJ, Juni P, Kastrati A, Koller A, Kristensen SD, Niebauer J, Richter DJ, Seferovic PM, Sibbing D, Stefanini GG, Windecker S, Yadav R, Zembala MO, Group ESCSD (2019) 2018 ESC/EACTS Guidelines on myocardial revascularization. Eur Heart J 40:87–165. https://doi.org/10.1093/eurheartj/ehy394
Ngkelo A, Richart A, Kirk JA, Bonnin P, Vilar J, Lemitre M, Marck P, Branchereau M, Le Gall S, Renault N, Guerin C, Ranek MJ, Kervadec A, Danelli L, Gautier G, Blank U, Launay P, Camerer E, Bruneval P, Menasche P, Heymes C, Luche E, Casteilla L, Cousin B, Rodewald HR, Kass DA, Silvestre JS (2016) Mast cells regulate myofilament calcium sensitization and heart function after myocardial infarction. J Exp Med 213:1353–1374. https://doi.org/10.1084/jem.20160081
Nossuli TO, Lakshminarayanan V, Baumgarten G, Taffet GE, Ballantyne CM, Michael LH, Entman ML (2000) A chronic mouse model of myocardial ischemia-reperfusion: essential in cytokine studies. Am J Physiol Heart Circ Physiol 278:H1049–H1055. https://doi.org/10.1152/ajpheart.2000.278.4.H1049
O’Connell TD, Swigart PM, Rodrigo MC, Ishizaka S, Joho S, Turnbull L, Tecott LH, Baker AJ, Foster E, Grossman W, Simpson PC (2006) Alpha1-adrenergic receptors prevent a maladaptive cardiac response to pressure overload. J Clin Invest 116:1005–1015. https://doi.org/10.1172/JCI22811
Oka T, Maillet M, Watt AJ, Schwartz RJ, Aronow BJ, Duncan SA, Molkentin JD (2006) Cardiac-specific deletion of Gata4 reveals its requirement for hypertrophy, compensation, and myocyte viability. Circ Res 98:837–845. https://doi.org/10.1161/01.RES.0000215985.18538.c4
Opavsky MA, Penninger J, Aitken K, Wen WH, Dawood F, Mak T, Liu P (1999) Susceptibility to myocarditis is dependent on the response of alphabeta T lymphocytes to coxsackieviral infection. Circ Res 85:551–558. https://doi.org/10.1161/01.RES.85.6.551
Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM (2006) Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med 355:251–259. https://doi.org/10.1056/NEJMoa052256
Oyama J, Blais C Jr, Liu X, Pu M, Kobzik L, Kelly RA, Bourcier T (2004) Reduced myocardial ischemia-reperfusion injury in toll-like receptor 4-deficient mice. Circulation 109:784–789. https://doi.org/10.1161/01.CIR.0000112575.66565.84
Papageorgiou AP, Swinnen M, Vanhoutte D, VandenDriessche T, Chuah M, Lindner D, Verhesen W, de Vries B, D’Hooge J, Lutgens E, Westermann D, Carmeliet P, Heymans S (2012) Thrombospondin-2 prevents cardiac injury and dysfunction in viral myocarditis through the activation of regulatory T-cells. Cardiovasc Res 94:115–124. https://doi.org/10.1093/cvr/cvs077
Patel B, Bansal SS, Ismahil MA, Hamid T, Rokosh G, Mack M, Prabhu SD (2018) CCR2(+) monocyte-derived infiltrating macrophages are required for adverse cardiac remodeling during pressure overload. JACC Basic Transl Sci 3:230–244. https://doi.org/10.1016/j.jacbts.2017.12.006
Paul M, Poyan Mehr A, Kreutz R (2006) Physiology of local renin-angiotensin systems. Physiol Rev 86:747–803. https://doi.org/10.1152/physrev.00036.2005
Peng H, Yang XP, Carretero OA, Nakagawa P, D’Ambrosio M, Leung P, Xu J, Peterson EL, Gonzalez GE, Harding P, Rhaleb NE (2011) Angiotensin II-induced dilated cardiomyopathy in Balb/c but not C57BL/6 J mice. Exp Physiol 96:756–764. https://doi.org/10.1113/expphysiol.2011.057612
Pfeffer MA, Pfeffer JM, Fishbein MC, Fletcher PJ, Spadaro J, Kloner RA, Braunwald E (1979) Myocardial infarct size and ventricular function in rats. Circ Res 44:503–512. https://doi.org/10.1161/01.RES.44.4.503
Pinkert S, Westermann D, Wang X, Klingel K, Dorner A, Savvatis K, Grossl T, Krohn S, Tschope C, Zeichhardt H, Kotsch K, Weitmann K, Hoffmann W, Schultheiss HP, Spiller OB, Poller W, Fechner H (2009) Prevention of cardiac dysfunction in acute coxsackievirus B3 cardiomyopathy by inducible expression of a soluble coxsackievirus-adenovirus receptor. Circulation 120:2358–2366. https://doi.org/10.1161/CIRCULATIONAHA.108.845339
Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, Tallquist MD (2016) Revisiting cardiac cellular composition. Circ Res 118:400–409. https://doi.org/10.1161/CIRCRESAHA.115.307778
Ponta H, Sherman L, Herrlich PA (2003) CD44: from adhesion molecules to signalling regulators. Nat Rev Mol Cell Biol 4:33–45. https://doi.org/10.1038/nrm1004
Prabhu SD, Frangogiannis NG (2016) The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res 119:91–112. https://doi.org/10.1161/CIRCRESAHA.116.303577
Proudfoot AE, Handel TM, Johnson Z, Lau EK, LiWang P, Clark-Lewis I, Borlat F, Wells TN, Kosco-Vilbois MH (2003) Glycosaminoglycan binding and oligomerization are essential for the in vivo activity of certain chemokines. Proc Natl Acad Sci USA 100:1885–1890. https://doi.org/10.1073/pnas.0334864100
Rai V, Sharma P, Agrawal S, Agrawal DK (2017) Relevance of mouse models of cardiac fibrosis and hypertrophy in cardiac research. Mol Cell Biochem 424:123–145. https://doi.org/10.1007/s11010-016-2849-0
Regan JA, Mauro AG, Carbone S, Marchetti C, Gill R, Mezzaroma E, Valle Raleigh J, Salloum FN, Van Tassell BW, Abbate A, Toldo S (2015) A mouse model of heart failure with preserved ejection fraction due to chronic infusion of a low subpressor dose of angiotensin II. Am J Physiol Heart Circ Physiol 309:H771–H778. https://doi.org/10.1152/ajpheart.00282.2015
Ren G, Michael LH, Entman ML, Frangogiannis NG (2002) Morphological characteristics of the microvasculature in healing myocardial infarcts. J Histochem Cytochem 50:71–79. https://doi.org/10.1177/002215540205000108
Richards DA, Bao W, Rambo MV, Burgert M, Jucker BM, Lenhard SC (2013) Examining the relationship between exercise tolerance and isoproterenol-based cardiac reserve in murine models of heart failure. J Appl Physiol (1985) 114:1202–1210. https://doi.org/10.1152/japplphysiol.00556.2012
Rickard AJ, Morgan J, Bienvenu LA, Fletcher EK, Cranston GA, Shen JZ, Reichelt ME, Delbridge LM, Young MJ (2012) Cardiomyocyte mineralocorticoid receptors are essential for deoxycorticosterone/salt-mediated inflammation and cardiac fibrosis. Hypertension 60:1443–1450. https://doi.org/10.1161/HYPERTENSIONAHA.112.203158
Rickard AJ, Morgan J, Chrissobolis S, Miller AA, Sobey CG, Young MJ (2014) Endothelial cell mineralocorticoid receptors regulate deoxycorticosterone/salt-mediated cardiac remodeling and vascular reactivity but not blood pressure. Hypertension 63:1033–1040. https://doi.org/10.1161/HYPERTENSIONAHA.113.01803
Rickard AJ, Morgan J, Tesch G, Funder JW, Fuller PJ, Young MJ (2009) Deletion of mineralocorticoid receptors from macrophages protects against deoxycorticosterone/salt-induced cardiac fibrosis and increased blood pressure. Hypertension 54:537–543. https://doi.org/10.1161/hypertensionaha.109.131110
Rienks M, Carai P, Bitsch N, Schellings M, Vanhaverbeke M, Verjans J, Cuijpers I, Heymans S, Papageorgiou A (2017) Sema3A promotes the resolution of cardiac inflammation after myocardial infarction. Basic Res Cardiol 112:42. https://doi.org/10.1007/s00395-017-0630-5
Rienks M, Papageorgiou AP (2016) Novel regulators of cardiac inflammation: matricellular proteins expand their repertoire. J Mol Cell Cardiol 91:172–178. https://doi.org/10.1016/j.yjmcc.2016.01.008
Rifkin DB, Mazzieri R, Munger JS, Noguera I, Sung J (1999) Proteolytic control of growth factor availability. APMIS 107:80–85. https://doi.org/10.1111/j.1699-0463.1999.tb01529.x
Roberts BJ, Dragon JA, Moussawi M, Huber SA (2012) Sex-specific signaling through Toll-Like Receptors 2 and 4 contributes to survival outcome of Coxsackievirus B3 infection in C57Bl/6 mice. Biol Sex Differ 3:25. https://doi.org/10.1186/2042-6410-3-25
Roberts BJ, Moussawi M, Huber SA (2013) Sex differences in TLR2 and TLR4 expression and their effect on coxsackievirus-induced autoimmune myocarditis. Exp Mol Pathol 94:58–64. https://doi.org/10.1016/j.yexmp.2012.06.005
Rockman HA, Ross RS, Harris AN, Knowlton KU, Steinhelper ME, Field LJ, Ross J Jr, Chien KR (1991) Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc Natl Acad Sci USA 88:8277–8281. https://doi.org/10.1073/pnas.88.18.8277
Rose NR (2014) Learning from myocarditis: mimicry, chaos and black holes. F1000Prime Rep 6:25. https://doi.org/10.12703/p6-25
Rosin NL, Sopel MJ, Falkenham A, Lee TD, Legare JF (2015) Disruption of collagen homeostasis can reverse established age-related myocardial fibrosis. Am J Pathol 185:631–642. https://doi.org/10.1016/j.ajpath.2014.11.009
Salvador AM, Moss ME, Aronovitz M, Mueller KB, Blanton RM, Jaffe IZ, Alcaide P (2017) Endothelial mineralocorticoid receptor contributes to systolic dysfunction induced by pressure overload without modulating cardiac hypertrophy or inflammation. Physiol Rep. https://doi.org/10.14814/phy2.13313
Saxena A, Chen W, Su Y, Rai V, Uche OU, Li N, Frangogiannis NG (2013) IL-1 induces proinflammatory leukocyte infiltration and regulates fibroblast phenotype in the infarcted myocardium. J Immunol 191:4838–4848. https://doi.org/10.4049/jimmunol.1300725
Schellings MW, Vanhoutte D, Swinnen M, Cleutjens JP, Debets J, van Leeuwen RE, d’Hooge J, Van de Werf F, Carmeliet P, Pinto YM, Sage EH, Heymans S (2009) Absence of SPARC results in increased cardiac rupture and dysfunction after acute myocardial infarction. J Exp Med 206:113–123. https://doi.org/10.1084/jem.20081244
Schiller M, Javelaud D, Mauviel A (2004) TGF-beta-induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci 35:83–92. https://doi.org/10.1016/j.jdermsci.2003.12.006
Schloss MJ, Horckmans M, Nitz K, Duchene J, Drechsler M, Bidzhekov K, Scheiermann C, Weber C, Soehnlein O, Steffens S (2016) The time-of-day of myocardial infarction onset affects healing through oscillations in cardiac neutrophil recruitment. EMBO Mol Med 8:937–948. https://doi.org/10.15252/emmm.201506083
Schroder K, Tschopp J (2010) The inflammasomes. Cell 140:821–832. https://doi.org/10.1016/j.cell.2010.01.040
Schroen B, Heymans S, Sharma U, Blankesteijn WM, Pokharel S, Cleutjens JP, Porter JG, Evelo CT, Duisters R, van Leeuwen RE, Janssen BJ, Debets JJ, Smits JF, Daemen MJ, Crijns HJ, Bornstein P, Pinto YM (2004) Thrombospondin-2 is essential for myocardial matrix integrity: increased expression identifies failure-prone cardiac hypertrophy. Circ Res 95:515–522. https://doi.org/10.1161/01.RES.0000141019.20332.3e
Schultheiss HP, Kuhl U, Cooper LT (2011) The management of myocarditis. Eur Heart J 32:2616–2625. https://doi.org/10.1093/eurheartj/ehr165
Schultz JC, Hilliard AA, Cooper LT Jr, Rihal CS (2009) Diagnosis and treatment of viral myocarditis. Mayo Clin Proc 84:1001–1009. https://doi.org/10.1016/S0025-6196(11)60670-8
Schultz Jel J, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, Kimball TR, Doetschman T (2002) TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest 109:787–796. https://doi.org/10.1172/JCI14190
Serini G, Bochaton-Piallat ML, Ropraz P, Geinoz A, Borsi L, Zardi L, Gabbiani G (1998) The fibronectin domain ED-A is crucial for myofibroblastic phenotype induction by transforming growth factor-beta1. J Cell Biol 142:873–881. https://doi.org/10.1083/jcb.142.3.873
Shen JZ, Morgan J, Tesch GH, Rickard AJ, Chrissobolis S, Drummond GR, Fuller PJ, Young MJ (2016) Cardiac tissue injury and remodeling is dependent upon MR regulation of activation pathways in cardiac tissue macrophages. Endocrinology 157:3213–3223. https://doi.org/10.1210/en.2016-1040
Shen Y, Cheng F, Sharma M, Merkulova Y, Raithatha SA, Parkinson LG, Zhao H, Westendorf K, Bohunek L, Bozin T, Hsu I, Ang LS, Williams SJ, Bleackley RC, Eriksson JE, Seidman MA, McManus BM, Granville DJ (2016) Granzyme B deficiency protects against angiotensin II-induced cardiac fibrosis. Am J Pathol 186:87–100. https://doi.org/10.1016/j.ajpath.2015.09.010
Shi Y, Chen C, Lisewski U, Wrackmeyer U, Radke M, Westermann D, Sauter M, Tschope C, Poller W, Klingel K, Gotthardt M (2009) Cardiac deletion of the Coxsackievirus-adenovirus receptor abolishes Coxsackievirus B3 infection and prevents myocarditis in vivo. J Am Coll Cardiol 53:1219–1226. https://doi.org/10.1016/j.jacc.2008.10.064
Shi Y, Massague J (2003) Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113:685–700. https://doi.org/10.1016/S0092-8674(03)00432-X
Shinde AV, Frangogiannis NG (2014) Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol 70:74–82. https://doi.org/10.1016/j.yjmcc.2013.11.015
Silberman GA, Fan TH, Liu H, Jiao Z, Xiao HD, Lovelock JD, Boulden BM, Widder J, Fredd S, Bernstein KE, Wolska BM, Dikalov S, Harrison DG, Dudley SC Jr (2010) Uncoupled cardiac nitric oxide synthase mediates diastolic dysfunction. Circulation 121:519–528. https://doi.org/10.1161/CIRCULATIONAHA.109.883777
Singer AJ, Clark RA (1999) Cutaneous wound healing. N Engl J Med 341:738–746. https://doi.org/10.1056/NEJM199909023411006
Soehnlein O, Lindbom L (2010) Phagocyte partnership during the onset and resolution of inflammation. Nat Rev Immunol 10:427–439. https://doi.org/10.1038/nri2779
Sorokin L (2010) The impact of the extracellular matrix on inflammation. Nat Rev Immunol 10:712–723. https://doi.org/10.1038/nri2852
Sriramula S, Francis J (2015) Tumor necrosis factor—alpha is essential for angiotensin II-induced ventricular remodeling: role for oxidative stress. PLoS One 10:e0138372. https://doi.org/10.1371/journal.pone.0138372
Suryakumar G, Kasiganesan H, Balasubramanian S, Kuppuswamy D (2010) Lack of beta3 integrin signaling contributes to calpain-mediated myocardial cell loss in pressure-overloaded myocardium. J Cardiovasc Pharmacol 55:567–573. https://doi.org/10.1097/FJC.0b013e3181d9f5d4
Szabo Z, Magga J, Alakoski T, Ulvila J, Piuhola J, Vainio L, Kivirikko KI, Vuolteenaho O, Ruskoaho H, Lipson KE, Signore P, Kerkela R (2014) Connective tissue growth factor inhibition attenuates left ventricular remodeling and dysfunction in pressure overload-induced heart failure. Hypertension 63:1235–1240. https://doi.org/10.1161/HYPERTENSIONAHA.114.03279
Takeda K, Akira S (2005) Toll-like receptors in innate immunity. Int Immunol 17:1–14. https://doi.org/10.1093/intimm/dxh186
Takimoto E, Champion HC, Li M, Belardi D, Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y, Kass DA (2005) Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and reverses cardiac hypertrophy. Nat Med 11:214–222. https://doi.org/10.1038/nm1175
Tang TT, Yuan J, Zhu ZF, Zhang WC, Xiao H, Xia N, Yan XX, Nie SF, Liu J, Zhou SF, Li JJ, Yao R, Liao MY, Tu X, Liao YH, Cheng X (2012) Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res Cardiol 107:232. https://doi.org/10.1007/s00395-011-0232-6
Tank J, Lindner D, Wang X, Stroux A, Gilke L, Gast M, Zietsch C, Skurk C, Scheibenbogen C, Klingel K, Lassner D, Kuhl U, Schultheiss HP, Westermann D, Poller W (2014) Single-target RNA interference for the blockade of multiple interacting proinflammatory and profibrotic pathways in cardiac fibroblasts. J Mol Cell Cardiol 66:141–156. https://doi.org/10.1016/j.yjmcc.2013.11.004
Tesch GH, Young MJ (2017) Mineralocorticoid receptor signaling as a therapeutic target for renal and cardiac fibrosis. Front Pharmacol 8:313. https://doi.org/10.3389/fphar.2017.00313
Thygesen K, Alpert JS, Jaffe AS, Chaitman BR, Bax JJ, Morrow DA, White HD, Executive Group on behalf of the Joint European Society of Cardiology/American College of Cardiology/American Heart Association/World Heart Federation Task Force for the Universal Definition of Myocardial I (2018) Fourth universal definition of myocardial infarction (2018). Circulation 138:e618–e651. https://doi.org/10.1161/cir.0000000000000617
Tian Z, Zheng H, Li J, Li Y, Su H, Wang X (2012) Genetically induced moderate inhibition of the proteasome in cardiomyocytes exacerbates myocardial ischemia-reperfusion injury in mice. Circ Res 111:532–542. https://doi.org/10.1161/CIRCRESAHA.112.270983
Trueblood NA, Xie Z, Communal C, Sam F, Ngoy S, Liaw L, Jenkins AW, Wang J, Sawyer DB, Bing OH, Apstein CS, Colucci WS, Singh K (2001) Exaggerated left ventricular dilation and reduced collagen deposition after myocardial infarction in mice lacking osteopontin. Circ Res 88:1080–1087. https://doi.org/10.1161/hh1001.090842
Usher MG, Duan SZ, Ivaschenko CY, Frieler RA, Berger S, Schutz G, Lumeng CN, Mortensen RM (2010) Myeloid mineralocorticoid receptor controls macrophage polarization and cardiovascular hypertrophy and remodeling in mice. J Clin Invest 120:3350–3364. https://doi.org/10.1172/JCI41080
Valero-Munoz M, Backman W, Sam F (2017) Murine models of heart failure with preserved ejection fraction: a “fishing expedition”. JACC Basic Transl Sci 2:770–789. https://doi.org/10.1016/j.jacbts.2017.07.013
Valero-Munoz M, Li S, Wilson RM, Hulsmans M, Aprahamian T, Fuster JJ, Nahrendorf M, Scherer PE, Sam F (2016) Heart failure with preserved ejection fraction induces beiging in adipose tissue. Circ Heart Fail 9:e002724. https://doi.org/10.1161/CIRCHEARTFAILURE.115.002724
Virag JI, Murry CE (2003) Myofibroblast and endothelial cell proliferation during murine myocardial infarct repair. Am J Pathol 163:2433–2440. https://doi.org/10.1016/S0002-9440(10)63598-5
Wang N, Liang H, Zen K (2014) Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol 5:614. https://doi.org/10.3389/fimmu.2014.00614
Weathington NM, van Houwelingen AH, Noerager BD, Jackson PL, Kraneveld AD, Galin FS, Folkerts G, Nijkamp FP, Blalock JE (2006) A novel peptide CXCR ligand derived from extracellular matrix degradation during airway inflammation. Nat Med 12:317–323. https://doi.org/10.1038/nm1361
Weber KT (1989) Cardiac interstitium in health and disease: the fibrillar collagen network. J Am Coll Cardiol 13:1637–1652. https://doi.org/10.1016/0735-1097(89)90360-4
Weinberg SE, Sena LA, Chandel NS (2015) Mitochondria in the regulation of innate and adaptive immunity. Immunity 42:406–417. https://doi.org/10.1016/j.immuni.2015.02.002
Weinzierl AO, Szalay G, Wolburg H, Sauter M, Rammensee HG, Kandolf R, Stevanovic S, Klingel K (2008) Effective chemokine secretion by dendritic cells and expansion of cross-presenting CD4-/CD8 + dendritic cells define a protective phenotype in the mouse model of coxsackievirus myocarditis. J Virol 82:8149–8160. https://doi.org/10.1128/JVI.00047-08
Weithauser A, Rauch U (2014) Role of protease-activated receptors for the innate immune response of the heart. Trends Cardiovasc Med 24:249–255. https://doi.org/10.1016/j.tcm.2014.06.004
Wessely R, Klingel K, Knowlton KU, Kandolf R (2001) Cardioselective infection with coxsackievirus B3 requires intact type I interferon signaling: implications for mortality and early viral replication. Circulation 103:756–761. https://doi.org/10.1161/01.cir.103.5.756
Wessely R, Klingel K, Santana LF, Dalton N, Hongo M, Jonathan Lederer W, Kandolf R, Knowlton KU (1998) Transgenic expression of replication-restricted enteroviral genomes in heart muscle induces defective excitation-contraction coupling and dilated cardiomyopathy. J Clin Invest 102:1444–1453. https://doi.org/10.1172/JCI1972
Westermann D, Becher PM, Lindner D, Savvatis K, Xia Y, Frohlich M, Hoffmann S, Schultheiss HP, Tschope C (2012) Selective PDE5A inhibition with sildenafil rescues left ventricular dysfunction, inflammatory immune response and cardiac remodeling in angiotensin II-induced heart failure in vivo. Basic Res Cardiol 107:308. https://doi.org/10.1007/s00395-012-0308-y
Westermann D, Heymans S (2014) Fibrosis or hypertrophy: let TIMPs decide. Cardiovasc Res 103:196–197. https://doi.org/10.1093/cvr/cvu160
Westermann D, Mersmann J, Melchior A, Freudenberger T, Petrik C, Schaefer L, Lullmann-Rauch R, Lettau O, Jacoby C, Schrader J, Brand-Herrmann SM, Young MF, Schultheiss HP, Levkau B, Baba HA, Unger T, Zacharowski K, Tschope C, Fischer JW (2008) Biglycan is required for adaptive remodeling after myocardial infarction. Circulation 117:1269–1276. https://doi.org/10.1161/CIRCULATIONAHA.107.714147
Westermann D, Savvatis K, Lindner D, Zietsch C, Becher PM, Hammer E, Heimesaat MM, Bereswill S, Volker U, Escher F, Riad A, Plendl J, Klingel K, Poller W, Schultheiss HP, Tschope C (2011) Reduced degradation of the chemokine MCP-3 by matrix metalloproteinase-2 exacerbates myocardial inflammation in experimental viral cardiomyopathy. Circulation 124:2082–2093. https://doi.org/10.1161/CIRCULATIONAHA.111.035964
Westermann D, Savvatis K, Schultheiss HP, Tschope C (2010) Immunomodulation and matrix metalloproteinases in viral myocarditis. J Mol Cell Cardiol 48:468–473. https://doi.org/10.1016/j.yjmcc.2009.08.019
Willems IE, Havenith MG, De Mey JG, Daemen MJ (1994) The alpha-smooth muscle actin-positive cells in healing human myocardial scars. Am J Pathol 145:868–875
Witt H, Schubert C, Jaekel J, Fliegner D, Penkalla A, Tiemann K, Stypmann J, Roepcke S, Brokat S, Mahmoodzadeh S, Brozova E, Davidson MM, Ruiz Noppinger P, Grohe C, Regitz-Zagrosek V (2008) Sex-specific pathways in early cardiac response to pressure overload in mice. J Mol Med (Berl) 86:1013–1024. https://doi.org/10.1007/s00109-008-0385-4
Woodall MC, Woodall BP, Gao E, Yuan A, Koch WJ (2016) Cardiac fibroblast GRK2 deletion enhances contractility and remodeling following ischemia/reperfusion injury. Circ Res 119:1116–1127. https://doi.org/10.1161/CIRCRESAHA.116.309538
Woodiwiss AJ, Tsotetsi OJ, Sprott S, Lancaster EJ, Mela T, Chung ES, Meyer TE, Norton GR (2001) Reduction in myocardial collagen cross-linking parallels left ventricular dilatation in rat models of systolic chamber dysfunction. Circulation 103:155–160. https://doi.org/10.1161/01.cir.103.1.155
Xia Y, Lee K, Li N, Corbett D, Mendoza L, Frangogiannis NG (2009) Characterization of the inflammatory and fibrotic response in a mouse model of cardiac pressure overload. Histochem Cell Biol 131:471–481. https://doi.org/10.1007/s00418-008-0541-5
Xiao H, Li H, Wang JJ, Zhang JS, Shen J, An XB, Zhang CC, Wu JM, Song Y, Wang XY, Yu HY, Deng XN, Li ZJ, Xu M, Lu ZZ, Du J, Gao W, Zhang AH, Feng Y, Zhang YY (2018) IL-18 cleavage triggers cardiac inflammation and fibrosis upon beta-adrenergic insult. Eur Heart J 39:60–69. https://doi.org/10.1093/eurheartj/ehx261
Xiong D, Yajima T, Lim BK, Stenbit A, Dublin A, Dalton ND, Summers-Torres D, Molkentin JD, Duplain H, Wessely R, Chen J, Knowlton KU (2007) Inducible cardiac-restricted expression of enteroviral protease 2A is sufficient to induce dilated cardiomyopathy. Circulation 115:94–102. https://doi.org/10.1161/CIRCULATIONAHA.106.631093
Xu S, Zhi H, Hou X, Cohen RA, Jiang B (2011) IkappaBbeta attenuates angiotensin II-induced cardiovascular inflammation and fibrosis in mice. Hypertension 58:310–316. https://doi.org/10.1161/HYPERTENSIONAHA.111.172031
Yajima T, Knowlton KU (2009) Viral myocarditis: from the perspective of the virus. Circulation 119:2615–2624. https://doi.org/10.1161/CIRCULATIONAHA.108.766022
Yan X, Anzai A, Katsumata Y, Matsuhashi T, Ito K, Endo J, Yamamoto T, Takeshima A, Shinmura K, Shen W, Fukuda K, Sano M (2013) Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J Mol Cell Cardiol 62:24–35. https://doi.org/10.1016/j.yjmcc.2013.04.023
Yang F, Liu YH, Yang XP, Xu J, Kapke A, Carretero OA (2002) Myocardial infarction and cardiac remodelling in mice. Exp Physiol 87:547–555. https://doi.org/10.1113/eph8702385
Yang XP, Liu YH, Scicli GM, Webb CR, Carretero OA (1997) Role of kinins in the cardioprotective effect of preconditioning: study of myocardial ischemia/reperfusion injury in B2 kinin receptor knockout mice and kininogen-deficient rats. Hypertension 30:735–740. https://doi.org/10.1161/01.HYP.30.3.735
Yu Q, Stamenkovic I (2000) Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev 14:163–176. https://doi.org/10.1101/gad.14.2.163
Zamilpa R, Zhang J, Chiao YA, de Castro Bras LE, Halade GV, Ma Y, Hacker SO, Lindsey ML (2013) Cardiac wound healing post-myocardial infarction: a novel method to target extracellular matrix remodeling in the left ventricle. Methods Mol Biol 1037:313–324. https://doi.org/10.1007/978-1-62703-505-7_18
Zaprutko J, Michalak M, Nowicka A, Dankowski R, Drozdz J, Ponikowski P, Opolski G, Nessler J, Nowalany-Kozielska E, Szyszka A (2017) Hospitalisation length and prognosis in heart failure patients. Kardiol Pol 75:323–331. https://doi.org/10.5603/KP.a2016.0183
Zhou P, Pu WT (2016) Recounting cardiac cellular composition. Circ Res 118:368–370. https://doi.org/10.1161/CIRCRESAHA.116.308139
Zhu M, Goetsch SC, Wang Z, Luo R, Hill JA, Schneider J, Morris SM Jr, Liu ZP (2015) FoxO4 promotes early inflammatory response upon myocardial infarction via endothelial Arg1. Circ Res 117:967–977. https://doi.org/10.1161/CIRCRESAHA.115.306919
Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I (2004) Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol 6:499–506. https://doi.org/10.1038/ncb1137
Zymek P, Bujak M, Chatila K, Cieslak A, Thakker G, Entman ML, Frangogiannis NG (2006) The role of platelet-derived growth factor signaling in healing myocardial infarcts. J Am Coll Cardiol 48:2315–2323. https://doi.org/10.1016/j.jacc.2006.07.060
Acknowledgements
The authors thank Katharina Scherschel for excellent help and support in microscopic imaging techniques.
Funding
This work was funded by the German Ministry of Research and Education (DZHK, German Center of Cardiovascular Research), Grant number 81365-150.
Author information
Authors and Affiliations
Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work and approved the manuscript for publication.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Bacmeister, L., Schwarzl, M., Warnke, S. et al. Inflammation and fibrosis in murine models of heart failure. Basic Res Cardiol 114, 19 (2019). https://doi.org/10.1007/s00395-019-0722-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-019-0722-5