
Abstract
Although ceramide accumulation in the heart is considered a major factor in promoting apoptosis and cardiac disorders, including heart failure, lipotoxicity and ischemia–reperfusion injury, little is known about ceramide’s role in mediating changes in contractility. In the present study, we measured the functional consequences of acute exposure of isolated field-stimulated adult rat cardiomyocytes to C6-ceramide. Exogenous ceramide treatment depressed the peak amplitude and the maximal velocity of shortening without altering intracellular calcium levels or kinetics. The inactive ceramide analog C6-dihydroceramide had no effect on myocyte shortening or [Ca2+]i transients. Experiments testing a potential role for C6-ceramide-mediated effects on activation of protein kinase C (PKC) demonstrated evidence for signaling through the calcium-independent isoform, PKCε. We employed 2-dimensional electrophoresis and anti-phospho-peptide antibodies to test whether treatment of the cardiomyocytes with C6-ceramide altered myocyte shortening via PKC-dependent phosphorylation of myofilament proteins. Compared to controls, myocytes treated with ceramide exhibited increased phosphorylation of myosin binding protein-C (cMyBP-C), specifically at Ser273 and Ser302, and troponin I (cTnI) at sites apart from Ser23/24, which could be attenuated with PKC inhibition. We conclude that the altered myofilament response to calcium resulting from multiple sites of PKC-dependent phosphorylation contributes to contractile dysfunction that is associated with cardiac diseases in which elevations in ceramides are present.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
There is emerging evidence that lipid accumulation within the myocardium is a mechanism in heart failure associated with obesity and diabetes mellitus. Ceramide, which has been deemed a cardiotoxin, is elevated in animal models of obesity [7, 40, 67] as well as in heart failure patients [8]. In addition, studies have identified ceramide as a significant contributor to the development of dilated cardiomyopathy in animal models of lipotoxicity [40], as well as to the worsening of contractile impairment in the progressively failing heart under high levels of circulating fatty acids [41]. Despite these findings, surprisingly little is known about ceramide’s effects in directly altering cardiac contractility.
Our current knowledge of ceramide’s ability to alter myocellular mechanics is based on studies using exogenous application of the short-chain analog C2-ceramide to isolated rat cardiomyocytes. These studies reported a positive inotropic response to ceramide treatment, which was associated with both alterations in calcium fluxes and the sensitivity of the myofilaments to calcium [32, 46]. However, these studies were unable to determine the mechanism underlying such alterations. Moreover, treatment of cells with C2-ceramide has been shown to produce effects that are either less potent than endogenous ceramide [19] or non-specific due to its highly hydrophilic nature [60]. C6-ceramide, which mimics endogenous ceramide generation [14], is now recognized as a more physiologically relevant means to assess the effects of ceramide in vitro. Studies in skeletal muscle using either C6-ceramide or endogenously generated ceramide have shown reductions in maximal force production [14, 15], findings that are more consistent with the depressed contractile force typically associated with ceramide accumulation.
Our goal in the present study was to determine the role of ceramide in modulating myocyte contractility through use of C6-ceramide. Furthermore, we sought to define the mechanism by which ceramide alters contractility. To do this we treated isolated adult rat ventricular cardiomyocytes with C6-ceramide and assessed shortening mechanics and [Ca2+]i transients, as well as post-translational modifications of the major myofilament proteins. Our findings demonstrate that acute treatment of isolated cardiomyocytes with C6-ceramide induces a negative inotropic response through activation of PKCε. Additionally, assessment of the post-translational state of the myofilaments following ceramide treatment showed significant increases in PKC-mediated phosphorylation of cardiac troponin I (cTnI) and cardiac myosin binding protein-C (cMyBP-C).
Materials and methods
Animals
Adult male Sprague–Dawley rats between 180 and 200 g were used for cell isolation experiments. All animal procedures were conducted according to the guidelines instituted by the Animal Care and Use Committee at the University of Illinois at Chicago and the National Institutes of Health.
Isolation of rat ventricular myocytes
Myocytes were isolated as previously described [62]. Briefly, rats were heparinized (5000 U/kg body weight) and then anesthetized with 100 mg/kg body weight of pentobarbital sodium, intraperitoneally. Animals were assessed for proper depth of anesthesia and then hearts were quickly excised and cannulated via the ascending aorta. Hearts were perfused at 37 °C for 3 min with a bovine serum albumin (1 mg/mL)-added calcium-free control solution (BSA-CS) of the following composition (in mM): NaCl 133.5, KCl 4.0, NaH2PO4 1.2, HEPES 10.0, MgSO4 1.2, glucose 11.1; pH 7.4 with NaOH. Cells were then perfused with oxygenated BSA-CS containing 0.25 mg/mL type II collagenase (Worthington) and 0.03 mg/mL protease (Sigma-Aldrich) for 15–20 min. After digestion, the ventricles were removed, and dissociated by trituration for 10 min at 37 °C in BSA-CS + 50 μM calcium. The cell suspension was filtered through a mesh collector and cells were resuspended in fresh BSA-CS containing first 100 μM calcium, then 200, 500 μM, and finally 1 mM calcium. The cells were stored at room temperature (22–23 °C) and used immediately, through to 4 h post-isolation.
Experimental solutions
In experiments measuring intact cell shortening and [Ca2+]i transients, cells were perfused with CS (baseline) or CS with the addition of either DMSO (0.05 %), 1.25–10 μM of the cell-permeable ceramide analog C6-ceramide (N-hexanoyl-d-erythro-sphingosine, Sigma-Aldrich), 5 μM C6-dihydroceramide (Avanti Lipids), 1 μM Chelerythrine chloride (Sigma-Aldrich), 5 μM Go6976 (Tocris), or 5 μM Go6983.
Measurements of intact cell shortening and calcium transients
Cells were loaded at room temperature with 3 μM fura 2-AM (Invitrogen, stock in DMSO) diluted in CS-BSA with 1 mM Ca2+, for 10 min in the perfusion chamber. Following loading, de-esterification of the fura 2 indicator was accomplished with 10 min perfusion using a dye-free CS + 1.5 mM Ca2+ solution. Cells were field stimulated at 0.5 Hz and intracellular calcium ([Ca2+]i) transients and unloaded cell shortening were recorded simultaneously as previously described [53]. Recordings were stored in an acquisition software program (Felix32, Photon Technology International) for later analysis offline. [Ca2+]i was measured as a function of the ratio of 340/380 wavelength emission after subtraction of the background fluorescence. [Ca2+]i transient decay times (τ decay, ms) were evaluated by a monoexponential fit to the declining phase of the [Ca2+]i transient. After baseline (control) measurements, the perfusion solution was changed to the appropriate treatment solution and recordings were repeated at the appropriate time, as indicated in the figure legends.
Cell culture
Isolated cells were plated for 1 h in creatine–carnitine–taurine (CCT) medium (Medium 199, Cellgrow) on 60-mm dishes pre-coated with 10 µg/µL mouse laminin (Sigma-Aldrich). Plates were then washed twice with CCT medium to remove any dead cells and the medium was replaced with either the selected treatment medium or normal CCT (control). Cells were incubated for 5 min in the selected condition medium at 37 °C and then washed and collected in ice-cold buffer. Cell pellets were obtained following centrifugation at 12,000×g for 5 min.
Western blot
Cell pellets were lysed in UTC Buffer (8 M urea, 2 M thiourea, 4 % CHAPS) with sonication and stored at −80 °C for further Western blotting or 2-D DIGE (see below). Protein lysates (20 μg) from cells were separated on 12 % SDS gels followed by transfer of the protein to a 0.2 μm polyvinylidene difluoride (PVDF) membrane for Western blot analysis. Membranes were blocked and then re-incubated overnight at 4 °C in primary antibody, [phospho-cTnI (Ser23/24) (Cell Signaling), 1:400; cTnI (C5 clone, Fitzgerald) 1:5,000; phospho-Tm (Ser283) 1:10,000; Tm (CH-1, Iowa Hybridoma) 1:1,000; [cMyBP-C (C0-C1), 1:10,000; phospho-cMyBP-C (Ser273), 1:2,000; phospho-cMyBP-C (Ser282), 1:2,000; phospho-cMyBP-C (Ser302), 1,10,000; GAPDH (Santa Cruz), 1:1,000]. Membranes were washed for 30 min in tris-buffer saline with 0.1 % Tween-20 (TBS-T) and then incubated in secondary antibody [goat anti-mouse (Sigma-Aldrich) 1:40,000 for cTnI; donkey anti-rabbit (Santa Cruz) 1:20,000 for cMyBP-C; goat anti-rabbit (GE Healthcare) 1:20,000 for all others] conjugated to horseradish peroxidase for 1 h at room temperature. Membranes were washed for 30 min in TBS-T then developed by enhanced chemiluminescence on a ChemiDoc XRS + imager (BioRad). Band densities were analyzed using ImageLab software (BioRad).
2-Dimensional difference gel electrophoresis (2-D DIGE)
2-D DIGE was performed as previously described [26, 59]. Briefly, protein lysates (100 μg) were cleaned using a 2D Clean-up Kit (GE Healthcare) and the precipitated protein was resuspended in UTC buffer. Samples were then labeled with cyanine dyes (CyDye, GE Healthcare). 40 μg of sample was mixed with IEF buffer (8 M urea, 2 M thiourea, 4 % w/v Chaps, 10 mM EDTA pH 8.0, 250 mM DTT, plus ampholytes) and samples were isoelectrically focused on nonlinear 18 cm immobilized pH gradient (IPG) 4–7 or 7–11 pH strips (GE Healthcare) using the standard protocols. After focusing, samples were equilibrated in 1 % DTT (w/v) dissolved in IEF Equilibration buffer (IEF-EQ; 6 M urea, 5 % SDS (w/v), 30 % glycerol (v/v)), then in 2.5 % iodoacetamide (w/v) dissolved in IEF-EQ. The second dimension electrophoresis was run on a 12 % SDS resolving gel. Gels were imaged using a Typhoon 9410 Imager (GE Healthcare) with Cy3 (532-nm laser), Cy5 (633-nm laser) and Cy2 (488-nm laser). Images were analyzed using PDQuest version 8 advanced (Biorad).
Subcellular fractionation
All steps were carried out on ice or at 4 °C. Following culturing, cells were manually scraped using 120 μL of ice-cold fractionation buffer [FB; 20 mM Tris–HCl, 2 mM EDTA, 0.5 mM EGTA, 0.3 mM sucrose, pH 7.4 with mammalian protease inhibitor cocktail (Sigma, 1:100) and phosphatase inhibitor cocktail (Calbiochem 1:100)] and centrifuged for 5 min at 20,000×g to wash and collect cells. The supernatant was removed and cell pellets were resuspended in 30 μL of FB. Samples were then homogenized with a micro ground glass Duall homogenizer for 1 min (Fig. 1, Fraction a—whole cell homogenate) and centrifuged at 1,000×g for 5 min. The supernatant fraction was saved and the pellet re-homogenized 3 additional times with the final pellet (Fraction b) ultimately discarded. Supernatant fractions from each homogenization were pooled together (Fraction c—enriched sample). Cytosolic and particulate fractions were then separated by ultracentrifugation at 40,000×g for 30 min. The resulting supernatant fraction was kept as the cytosolic fraction (Fraction d—cytosolic) and the pellet was resuspended in sucrose-free FB with 1 % Triton, incubated for 1 h (Fraction e—particulate), then centrifuged at 80,000×g for 15 min. The resulting supernatant fraction was kept as the membrane fraction (Fraction f—membrane) and pellets were discarded (Fraction g). Prior to experimentation, successful fractionation of the cytosol and membrane was confirmed using Western blotting with antibodies against the cytosolic protein GAPDH and the membrane protein Na+/K+ ATPase (Millipore; 1:5,000) (Fig. 1). Protein samples (20 μg) of cytosolic and membrane fractions were separated by SDS-PAGE and protein was transferred to 0.2 μm nitrocellulose membrane. Membranes were stained with MemCode (Pierce Biotechnology) and imaged on a Chemidoc XRS + imager to quantitate total protein. After the stain was removed, membranes were then incubated overnight at 4 °C in primary antibody [PKCε (Millipore), 1:1,500; PKCδ (Santa Cruz), 1:500; PKCζ (Santa Cruz), 1:500] diluted in TBS-T with 2.5 % milk. Membranes were incubated in secondary antibody (goat anti-mouse (Sigma) 1:50,000 for Na+/K+ ATPase; goat anti-rabbit (GE Healthcare), 1:40,000 for all others) conjugated to horseradish peroxidase for 1 h at room temperature. Membranes were developed by enhanced chemiluminescence on the ChemiDoc XRS + imager. Band densities were analyzed using ImageLab software and normalized to the total protein density in each respective lane. Cytosolic and membrane fractions from each sample were expressed as the percentage of total (cytosolic + membrane).

Western blot verifying efficient subcellular fractionation. Representative western blot of GAPDH and Na+/K+ ATPase performed on samples a–g taken during each step of the subcellular fractionation protocol. Letters a–g represents each sample fraction taken during (see “Materials and methods”) the subcellular fractionation. The presence of GAPDH predominately within the cytosolic fraction and Na+/K+ ATPase only within the corresponding membrane fraction confirms proper subcellular fractionation
Statistical analysis
Values are presented as mean ± SEM. For measures of unloaded cell shortening and [Ca2+]i transients, where 3 groups were compared, statistical analyses were performed using one-way repeated measures ANOVA along with a multi-comparison Newman–Keuls post hoc test to make comparisons among groups. All other data were analyzed using a paired or unpaired Student’s t test, as appropriate. A level of p < 0.05 was considered significant throughout.
Results
Acute treatment of isolated cardiomyocytes with ceramide leads to a depression in contractility without altering Ca2+ transients
First we assessed the functional consequence of exogenous ceramide exposure in both a concentration- and a time-dependent manner. As depicted in Fig. 2a, b, treatment of isolated rat cardiomyocytes for 5 min with C6-ceramide decreased the peak amplitude of cell shortening in a concentration-dependent manner, with a maximal decrease of 25.1 ± 3.4 % at 5 μM concentration. Importantly, treatment with vehicle (0.05 % DMSO) had no effect on cell shortening. The ceramide-mediated decrease in peak amplitude of shortening was also time-dependent, as shown in Fig. 2c, d. Regardless of time or concentration studied, ceramide exposure had no effect on the peak Ca2+ transient or the rate of transient decline (τ), as assessed by the fura 2 ratio (Fig. 2a insert). Based on these initial studies, further characterization of the functional effects of ceramide on cell mechanics and intracellular [Ca2+] was conducted using 5 min exposure of myocytes to 5 μM C6-ceramide.

Concentration and time-dependent effects of exogenous C6-ceramide treatment on contractile mechanics and [Ca2+]i in ventricular cardiomyocytes. a Overlay of unloaded cell shortening measurements from a single cardiomyocyte under field stimulation at 0.5 Hz at baseline and during treatment with sequentially increasing concentrations of C6-ceramide. Inset, overlay of simultaneous Ca2+-transient measures corresponding to each cell shortening measure. b The quantified change in the percentage of cell shortening relative to baseline following treatment with each concentration of C6-ceramide. *p < 0.01 vs. baseline; n = 6. c Representative cell shortening measurements taken under steady-state pacing at baseline conditions and following 30 s, 2 and 5 min of 5 μM C6-ceramide treatment. d The quantified change in the percentage of cell shortening relative to baseline during the time course for 5 μM C6-ceramide treatment. *p < 0.05 vs. baseline, # p < 0.01 vs. 0.5 min, n = 8
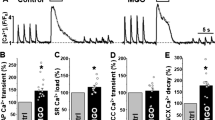
Figure 3a shows an overlay of cell shortening and [Ca2+]i transient at baseline (control) and after ceramide treatment. In these experiments, the average resting cell length at baseline was 108.2 ± 7.5 μm and did not differ with ceramide treatment. Acute exposure of isolated cardiomyocytes to 5 μM C6-ceramide resulted in a significant depression in the peak shortening amplitude (baseline = 8.8 ± 0.8 % vs. ceramide = 7.3 ± 0.7 %). The peak rate of contraction was also reduced following ceramide treatment (baseline = 86.0 ± 7.9 μm/s vs. ceramide = 72.7 ± 6.6 μm/s), as was the peak rate of relaxation (baseline = 56.7 ± 4.2 μm/s vs. ceramide = 50.8 ± 3.4 μm/s), although to a lesser extent (Fig. 3b). These changes in cellular mechanics following ceramide treatment occurred independently of changes in [Ca2+]i, demonstrated by the lack of an effect of ceramide treatment on peak [Ca2+]i amplitude (Δ fura ratio; baseline = 0.34 ± 0.06 vs. ceramide = 0.36 ± 0.07), the decay time constant, τ (baseline = 281.6 ± 23.9 ms vs. ceramide = 290.0 ± 23.6 ms) (Fig. 3c) and the resting [Ca2+]i (baseline = 0.31 ± 0.03 vs. ceramide = 0.31 ± 0.03). Table 1 shows the effects of ceramide, along with all treatments used, on the parameters of cell shortening and [Ca2+]i expressed as a percentage of baseline measures.

Effects of acute C6-ceramide treatment on contractile mechanics and [Ca2+]i in ventricular cardiomyocytes. a Overlay of representative unloaded cell shortening measurements with simultaneous Ca2+ transient measurements in a single cardiomyocyte under field stimulation (0.5 Hz) at baseline and after exogenous treatment with 5 μM C6-ceramide. b Quantification of the contractile parameters (percent of shortening and maximal rates of contraction and relaxation) and c Ca2+ transient parameters (peak amplitude of the fura 2 ratio and the time constant, τ, for the declining phase of the transient). Statistics were done using a paired Student’s t test. *p < 0.05 vs. baseline, **p < 0.001 vs. baseline, n = 8
To test the specificity of ceramide’s functional effect, we exposed cardiomyocytes to C6-dihydroceramide, an inactive ceramide analog [2], or 0.05 % DMSO (the concentration of DMSO present in the ceramide and dihydroceramide perfusion buffers) and measured cell shortening and [Ca2+]i transients. As shown in Fig. 4, both DMSO and dihydroceramide failed to alter any of the parameters of cell shortening or [Ca2+]i transients. These findings indicate that the negative inotropic response induced by ceramide is specific to ceramide’s known bioactivity. Moreover, the negative inotropic response to ceramide treatment in the presence of unaltered [Ca2+]i suggests downstream alterations at the level of the myofilament.

Assessment of functional specificity using the non-bioactive sphingolipid C6-dihydroceramide. a Quantification of contractile parameters (percent of shortening and maximal rates of contraction and relaxation) before treatment, after vehicle treatment (0.05 % DMSO) and following treatment with 5 μM C6-dihydroceramide. b Quantification of Ca2+ transient parameters (peak amplitude of the fura 2 ratio (340 nm/380 nm) and the time constant, τ, for the declining phase of the transient) before treatment, after vehicle treatment and following treatment with 5 μM C6-dihydroceramide. n = 5
PKCε mediates ceramide’s negative inotropic response
To address whether PKC is involved in mediating ceramide’s effects on cardiomyocyte function we repeated measures of intact cell shortening and [Ca2+]i transients in isolated myocytes in the presence of ceramide + the general PKC inhibitor chelerythrine chloride. Cardiomyocyte contractile parameters were unchanged from baseline levels when exposed to ceramide in the presence of chelerythrine chloride (Fig. 5a), implying that ceramide signals through PKCs to depress cardiomyocyte contractility. The peak shortening amplitude was 8.1 ± 1.8, 7.8 ± 1.8 and 7.9 ± 1.9 % for baseline, chelerythrine chloride treatment alone and ceramide + chelerythrine chloride treatment, respectively. Maximal shortening velocity was 82.9 ± 15.3, 80.3.7 ± 15.4, 81.2 ± 15.4 μm/s and maximal relengthening velocity was 50.3 ± 11.5, 48.1 ± 11.5, and 49.1 ± 11.4 μm/s for baseline, chelerythrine chloride treatment alone and ceramide + chelerythrine chloride treatment, respectively. Measurements of [Ca2+]i transients were also unchanged among all groups (Table 1).

Attenuation of the functional effects of acute C6-ceramide treatment by inhibition of PKCs. a Quantification of contractile parameters (percent of shortening and maximal rates of contraction and relaxation) at baseline and following treatment with 1 μM chelerythrine chloride and 1 μM chelerythrine chloride + 5 μM C6-ceramide. n = 6. b Quantification of contractile parameters (percent of shortening and maximal rates of contraction and relaxation) at baseline and following treatment with 5 μM Go6976 and 5 μM Go6976 + 5 μM C6-ceramide. *p < 0.01 vs. baseline, n = 6
It is known that rat cardiomyocytes express 4 isoforms of PKC, α, δ, ε and ζ [42]. Of these, isoform α is of the conventional class of PKCs and is dependent on calcium and diacylglycerol (DAG) for activation, while the other isoforms (novel PKCs, δ and ε; atypical PKC, ζ) are calcium independent [33]. Through use of the conventional PKC inhibitor, Go6976, we were able to demonstrate that ceramide mediates its negative inotropic effects through one of the calcium-independent isoforms of PKC (Fig. 5b). Treatment of cardiomyocytes with Go6976 alone had no effect on cell shortening parameters (peak shortening = 8.3 ± 0.5 % vs. baseline = 8.6 ± 0.4 %, velocity of shortening = 90.8 ± 10.9 μm/s vs. baseline = 90.8 ± 9.6 μm/s, velocity of relengthening = 64.0 ± 10.8 μm/s vs. baseline = 62.4 ± 8.9 μm/s) or [Ca2+]i transients (peak fura amplitude = 0.44 ± 0.07 vs. baseline = 0.43 ± 0.06, decay time constant (τ) = 234.6 ± 16.0 ms vs. baseline = 228.5 ± 15.2 ms). Moreover, Go6976 was unable to block ceramide’s functional effects when cells were exposed to Go6976 + ceramide. This is observed by ceramide’s ability to depress peak shortening amplitude (7.2 ± 0.5 % vs. baseline = 8.6 ± 0.4 %) and the maximal velocity of shortening (74.3 ± 10.4 μm/s vs. baseline = 90.8 ± 9.6 μm/s) with a reduced, but insignificant, change in the relengthening velocity (ceramide + Go6976 = 54.2 ± 9.0 μm/s vs. baseline = 62.4 ± 8.9 μm/s). Additionally, in the presence of Go6976 neither the [Ca2+]i transient (peak fura 2 amplitude for ceramide + Go6976 = 0.41 ± 0.06 vs. baseline = 0.43 ± 0.06), or decay time constant (τ) (ceramide + Go6976 = 243.6 ± 17.8 vs. baseline = 228.5 ± 15.2 ms) was unaltered among the treatment groups. These data were confirmed with use of G06983, another inhibitor of PKC which is selective for the α, β,δ, and γ isoforms [64] (see Table 1).
Subcellular fractionation and immunoblotting of the calcium-independent PKC isoforms were used as a reliable means to indirectly measure PKC activation through translocation. To do this, cardiomyocytes were fractionated into cytosolic and membrane compartments and the relative amount of each calcium-independent PKC isoform within that compartment was then determined using Western blotting. Cardiomyocytes were also treated with 100 nM of phorbol-12-myristate-13-acetate (PMA), a potent activator of conventional and novel PKCs, as a positive control, for PKCδ and ε. As expected, treatment of cardiomyocytes with 100 nM PMA resulted in translocation of virtually all of PKCε and PKCδ into the membrane fraction (98.60 ± 0.58 and 91.50 ± 0.05 %, respectively, n = 3), while the subcellular distribution of PKCζ remained unchanged by PMA treatment (membrane fraction = 33.60 ± 2.92 % vs. cytosolic fraction = 66.43 ± 2.92 %, n = 3) (Fig. 6a). Interestingly, ceramide treatment resulted in a significant shift in the subcellular distribution of PKCε, with 28.80 ± 3.80 % localized within the membrane fraction compared to 16.67 ± 1.90 % in untreated (control) samples. The distribution of PKCδ (membrane fraction = 55.32 ± 3.60 % in ceramide treated vs. 63.01 ± 2.60 % in untreated controls) and PKCζ (membrane fraction = 20.23 ± 6.72 % in ceramide treated vs. 37.53 ± 8.51 % in untreated controls) within the myocytes remained unchanged by ceramide treatment (Fig. 6b).

Subcellular fractionation of ventricular cardiomyocytes treated with C6-ceramide to assess translocation of the calcium-independent PKC isoforms. a Representative Western blots of the calcium-independent PKC isoforms in the cytosolic and membrane subcellular fractions of isolated cardiomyocytes in control cells, cells treated with 100 nM PMA, and cells treated with 10 μM C6-ceramide. b Quantification of the cytosolic and membrane subcellular fractions from control and C6-ceramide-treated samples. White bars represent the amount of PKC in the cytosolic fraction and grey bars represent the amount of PKC within the membrane fraction. Each band density was normalized to the total protein within that lane. *p < 0.05 vs. control, n = 3
Ceramide treatment results in increased phosphorylation of cTnI and cMyBP-C
Acute changes in cardiomyocyte function, as observed here, are likely the result of transient post-translational protein modifications. Moreover, it is well known that such post-translational modifications of myofilament proteins lead to alterations in contractility [27, 54]. Therefore, we tested whether acute exposure of cardiomyocytes to ceramide, or ceramide in the presence of the PKC inhibitor chelerythrine chloride had an effect on the post-translational state of myofilament proteins using 2D-DIGE, which allows for analysis of the post-translational state of the sarcomeric proteome on a global level. Figure 7 shows a representative scans of Cy5-labeled untreated (control) sample, Cy2-labeled ceramide-treated sample and Cy3-labeled ceramide + chelerythrine chloride sample run within the same gel, along with the representative merged image for direct comparison. Spots associated with cTnI are labeled in the merged image, with a shift in the migration of phosphorylated cTnI spots toward the acidic region of the strip in an additive fashion (i.e. acidity of P4 > P3 > P2, etc.). Analysis of the samples showed an increase in the tris-phosphorylated (P3) spot alone in samples treated with ceramide (22.8 ± 1.4 % vs. control = 14.0 ± 2.2 %, n = 7), which was attenuated by PKC inhibition (15.1 ± 2.7 %). Further 2D-DIGE analysis of the other myofilament proteins showed no significant differences in the post-translational state of Tm, RLC or cTnT (Fig. 8).

The role of PKC in C6-ceramide-mediated effects on cTnI phosphorylation. cTnI, cardiac troponin I; P, phosphorylated cardiac troponin I. Top images of the cTnI region of interest from Cy5 labeled control samples (channel 1), Cy2 labeled samples treated with 10 μM C6-ceramide (channel 2) and Cy3 labeled samples treated with 10 μM C6-ceramide in the presence of 1 μM chelerythrine chloride (channel 3) and the merged image (channel 1 + 2 + 3). Samples were pooled together and focused on an 18 cm nonlinear IPG 7-11 pH strip followed by standard 12 % SDS-PAGE for the second dimension. Bottom quantitative results for cTnI phosphorylation spots 1–4 in which each spot density is shown relative to the total density for all cTnI spots. *p < 0.05 as indicated; n = 7

Assessment of C6-ceramide-mediated effects of myofilament protein phosphorylation for Tm, RLC, and cTnT by 2D-DIGE. Tm tropomyosin, RLC regulatory light chain, cTnT cardiac troponin T, P phosphorylation. a Representative gel images for the region of interest including Tm, RLC and cTnT are shown in a merged image of control samples labeled with Cy3 (pseudo-colored green) and C6-ceramide-treated samples labeled with Cy5 (pseudo-colored red). b Results from the quantification of phosphorylation sites from Tm, RLC and cTnT expressed as a percentage of all spot density for the protein of interest. *p < 0.05 vs. control; n = 7
Assessment of site-specific myofilament protein phosphorylation upon C6-ceramide treatment was also assessed using phospho-peptide antibodies, where available. We used phospho-peptide antibodies (gift from Dr. Sakthivel Sadayappan) against 3 of the known phosphorylation sites for cMyBP-C. Figure 9A shows a concentration-dependent increase in the intensity of the immunoblot signal for Ser-273 and Ser-302 without changes in the Ser-282 signal in ceramide-treated samples. Quantification of samples confirmed an increase in the phosphorylation state of Ser-273 (1.5-fold) and Ser-302 (1.6-fold) with no difference in Ser-282 phosphorylation (1.1-fold). Immunoblotting with anti-phospho-peptide antibodies was also carried out for cTnI at Ser23/24 and for Tm at Ser283 (gift from Dr. David Wieczorek). As shown in Fig. 9b and c, no difference in phosphorylation at these sites was observed in untreated vs. ceramide-treated cardiomyocytes.

Site-specific immunoblotting for cMyBP-C, Tm and cTnI phosphorylation. Tm tropomyosin, cTnI cardiac troponin I, MyBP-C myosin binding protein-C, Ser serine. a Left, representative images from western blots using cMyBP-C phospho-specific antibodies against Ser-273, Ser-282 and Ser-302 in untreated samples and in samples treated with increasing doses of C6-ceramide. Right, Quantification of cMyBP-C phosphorylation levels in isolated cells treated with and without 10 μM C6-ceramide at Ser-273, Ser-282 and Ser302. GAPDH was used as a loading control. n = 5. b Western blot quantification of isolated cells treated with and without 10 μM C6-ceramide using a phospho-Tm (Ser283) antibody. n = 5. c Western blot quantification of isolated cells treated with and without 10 μM C6-ceramide using a phospho-cTnI (Ser23/24) antibody. *p < 0.05 vs. control; n = 6
Previous studies have shown phosphorylation of cMyBP-C at Ser-273, Ser-282 and Ser-302 to be mediated by PKA, whereas only two of these sites, Ser-273 and Ser-302, are phosphorylated by PKC [34]. This, coupled with the observed increase in the phosphorylation state of cTnI at sites apart from Ser23/24 (putative PKA sites) strongly support the likely involvement of PKC in mediating ceramide’s negative inotropic effects.
Discussion
Although ceramide accumulation in the heart is known to adversely affect cardiac function, the present study demonstrates for the first time that ceramide directly alters cardiomyocyte contractility through modulation of the post-translational state of the myofilament proteins. Moreover, our data strongly support ceramide signaling to the myofilaments via PKCε (Fig. 10).

Schematic diagram of the mechanism by which ceramide leads to negative inotropy. Accumulation of ceramide within the cardiomyocyte results in activation of PKCε, leading to phosphorylation of the myofilament proteins cTnI and cMyBP-C. These post-translational modifications within the sarcomere alter the basal function and cause a reduction in the ability of the contractile machinery to generate force
Previous studies have concluded that depressions in contractility observed when ceramide levels are elevated are due primarily to cell death via ceramide-stimulated apoptosis. However, our functional observations in intact cardiomyocytes revealed that ceramide directly regulates contractile function demonstrated by a depression in shortening amplitude and velocity following ceramide exposure, which occurred despite unaltered [Ca2+]i levels or kinetics. Moreover, the potential ceramide-mediated effects on apoptosis and cell death are most likely not relevant inasmuch as these were acute experiments. Although the observed changes were modest (17–25 % reduction), they would be projected to reduce force production and power output (the product of force and velocity) in the intact myocardium and contribute significantly to depressed contractile function.
The mechanism by which ceramide-mediated activation of PKC might alter contractile mechanics is likely to involve both the allosteric regulation of crossbridges reacting with the thin filament and crossbridge dynamics independent of thin filament activation. PKC-dependent phosphorylation of the myofilaments in vitro has been shown to reduce Ca2+-activated actomyosin ATPase, myofilament Ca2+ sensitivity and cooperativity with observed depressions in the maximal tension generation [6, 25, 28, 35–39, 44, 56]. Functionally, these changes are associated with reduced force production and shortening velocity, both loaded and unloaded [31, 55], resulting in depressed power output [23]. While reductions in isometric tension generation can be attributed to an altered activation state of the myofilaments, previous studies demonstrate that shortening velocities at zero load are unaffected by myofilament activation [13, 18] and are more likely to depend on crossbridge cycling [24]. However, exactly how PKC-dependent phosphorylation of the myofilaments modifies crossbridge dynamics is unclear. Whereas earlier studies by Pyle et al. [45] identified PKC-mediated effects on crossbridge detachment rates, as demonstrated by increased tension cost (an estimate of the crossbridge detachment rate [4, 11] ) in transgenic mice expressing an unphosphorylatable mutant cTnI (cTnI-S43A/45A), more recent studies have shown unaltered tension costs and, therefore, unaltered crossbridge detachment rates in transgenic (TG) mice in which all 3 PKC sites were psuedophosphorylated by glutamic acid substitution [26]. The authors, instead, attributed the negative inotropic response in TG mice to a decreased rate of crossbridge reaction with thin filaments and a Ca2+-independent persistence of the active state. Despite the discrepancy, in general PKC-mediated phosphorylation has been shown to reduce crossbridge cycling rate [36, 37, 39] and would be expected to prolong the duty cycle, consequently reducing shortening kinetics during ejection. This is likely also to be true for the PKC-dependent effects we observed following ceramide treatment. Moreover, evidence from others indicates that the effects we observed in isolated cardiomyocytes may be more pronounced in the intact, auxotonically loaded heart [29], translating to a marked reduction in pressure development, decreased extent of shortening during ejection and overall impairment of systolic function.
An important and novel finding of our study was that ceramide’s negative inotropic response was due to PKC-mediated phosphorylation of the myofilament proteins cTnI and cMyBP-C. Our biochemical assessment revealed increases in the phosphorylation of cMyBP-C at the PKC sites upon ceramide exposure, as well as increases in the phosphorylated form (P3) of cTnI which was prevented by inhibition of PKC. Previous studies support the idea that the phosphorylated P3 spot likely contains phosphorylation of cTnI on Ser-43/45 [1, 50, 51, 66]. Specifically, it has been shown that the higher charged, more acidic cTnI species identified by isoelectric mobility in IEF gels includes phosphorylation on the PKC sites [51], while the vast majority of mono-phosphorylated (cTnI p ) and bis-phosphorylated (cTnI pp ) cTnI found in the basal state contain cTnI phosphorylated at Ser-23/24 [1, 50, 66].
Although it is clear that the net phosphorylation state of myofilament proteins is critical for determining the overall contractile effect in the intact heart, our data in isolated, unloaded cardiomyocytes support a dominate role for cTnI phosphorylation in mediating ceramide’s negative inotropic effects. Functionally, PKC-mediated phosphorylation of cTnI in vitro, specifically at Ser-43/45, has been shown to reduce isometric tension and the calcium-activated ATPase rate by promoting the blocked state of thin-filament activation [25, 39, 45], an effect which is likely to alter the economy of muscle contraction. Indeed, a recent study by Hinken et al. [23] showed PKC treatment of skinned-rat cardiomyocytes reduced force production and loaded shortening velocity resulting in depressed power output. Phosphorylation of cTnI was shown to play a dominate role in these responses. Consistent with that study are data presented here demonstrating reduced shortening amplitude and velocity due to alterations in the post-translational state of the myofilaments. In vivo phosphorylation of cTnI at Ser43/45 has been shown to play a main role in the contractile dysfunction and progression to failure associated with PKCε overexpression, demonstrated by rescue of the PKCε overexpressing mouse phenotype when endogenous cTnI was partially replaced with a non-phosphorylatable cTnI at Ser43/45 (S43/45A) [51]. Interestingly, a study using mice, which constitutively express pseudo-phosphorylated cTnI at Ser43/45 and Thr144 (cTnIPKC-P), showed that minimal changes (7 %) in cTnI phosphorylation at the PKC sites were sufficient to cause contractile dysfunction [26]. It is worth noting here that the observed increase in phosphorylation induced by ceramide treatment in the current study was about 8 %. The aforementioned study suggests that in the context of the whole heart this degree of modification would be sufficient to impair contractility.
Unexpectedly, our observed increase in cMyBP-C phosphorylation was not associated with an increase in the rate of contraction or relaxation, as has been previously described [9, 49, 58]. One explanation for this lack of increase in contraction and relaxation rates is that increases in phosphorylation of the PKC sites at Ser273 and Ser302 without changes in Ser-282, as shown here, may impact contractile mechanics differently than when all three sites on cMyBP-C are phosphorylated to the same extent. In fact, studies examining the role of cMyBP-C phosphorylation on contractile mechanics have focused solely on PKA-mediated phosphorylation of cMyBP-C, in which case all three sites, Ser273, Ser282 and Ser302, are phosphorylated. However, Sadayappan et al. [48] have shown that pseudo-phosphorylation of cMyBP-C at the PKC sites was not sufficient to rescue contractile dysfunction in the cMyBP-C null background, while Ser282 alone was. The authors suggest that phosphorylation of Ser282 is essential for cMyBP-C’s ability to regulate contractility and may be the major determinate of cMyBP-C phosphorylation-induced alterations. Moreover, Florea et al. [17] found that constitutive phosphorylation of PP1 I-1 at its PKC sites resulted in depressed systolic function that was associated, in part, with reduced phosphorylation of cMyBP-C at Ser282. Future studies examining the functional differences between PKA and PKC-mediated phosphorylation of cMyBP-C may shed light on more recent findings. A second explanation is that perhaps phosphorylation of cMyBP-C does little to change relengthening kinetics in unloaded cell measurements. The notion of a load-dependence of relaxation under auxotonic conditions has long been established [5]. Moreover, in the unphosphorylated state cMyBP-C has been proposed to conform to a ‘viscous-load model’ in which it produces an opposing drag force through its ability to bind actin. This effect is reduced when phosphorylation of cMyBP-C blunts its binding to actin, allowing for less force-opposition and faster contraction and relaxation kinetics [61]. In this case, the influence of cMyBP-C phosphorylation may be reduced in the context of unloaded cardiomyocytes used here, particularly in terms of relengthening and may, therefore, account for our lack of change in relengthening velocity following ceramide treatment, despite an increase in cMyBP-C phosphorylation.
Given that measurements were taken in isolated cardiomyocytes at room temperature (22–24 °C) and not at physiologic temperatures of 37 °C, we cannot completely rule out the possibility of altered effects of ceramide at body temperature. Reductions in temperature have been shown to reduce the relative contribution of SR Ca2 + release to inotropy [52], as well as accelerate relaxation by both increasing calcium removal from the cytosol [43] and by reducing myofilament calcium sensitivity [21]. The latter may partially explain why increased calcium sensitivity owing to MyBP-C phosphorylation was not observed in this study.
Beyond the acute effects shown here, ceramide-induced activation of PKC may also be of significance in cardiac remodeling and the development of dilated cardiomyopathy associated with long-term elevations in ceramide [16, 40, 65, 67]. Activation of PKCs directly by ceramide has been shown to occur in the heart, leading to both insulin resistance [8] and reduced β-adrenergic signaling [12]. Likewise, it is well known that PKC activation induces hypertrophic growth within the heart [3, 47, 57, 63] and may further play a role in modifying actin capping dynamics [22] to promote addition of new sarcomeres into the myofilament lattice. Thus, we speculate that chronic elevations in ceramide may be one of the mechanistic triggers of cardiac decompensation, which leads to contractile dysfunction, ventricular dilation and the progression to failure. This hypothesis is supported by studies from Park et al. [40] who found that inhibition of de novo ceramide biosynthesis in LpLGPI overexpressing mice, which develop lipotoxic-induced dilated cardiomyopathy, resulted in reduced apoptosis, enhanced myocardial energetics and improved overall systolic function and survival. Interestingly, in a rat model of metabolic syndrome, elevations in TAG and long-chain acyl CoAs (ceramide content was not measured) caused membrane translocation and activation of PKCε leading to contractile dysfunction [10]. Nevertheless, future studies examining ceramide accumulation in human diseases of cardiac lipotoxicity will be critical for establishing a role for ceramide as a lipotoxic mediator in human pathology.
Although novel, these findings are limited by the use of exogenous treatment of a non-biological ceramide analog. The shorter chain length differs from endogenously produced ceramide, which is composed of a sphingoid base with an acyl chain length ranging from 14 to 28 carbons, the most abundant of which C20:0 and C24:0 in the heart [30]. Nevertheless, it has been demonstrated that 1–20 µM of short-chain ceramide analog administered exogenously to cells results in a cellular accumulation of ceramide that is within the range of endogenous levels and which elicits specific biological responses [20]. Moreover, in vitro use of C6-ceramide has been shown to promote negative inotropic responses similar to endogenously generated ceramide [15]. In the failing human heart, 1.5-fold elevations in total ceramide content have been shown [8]. This increase was associated with higher insulin resistance in heart failure patients, mediated by activation of PKCs. Interestingly, implantation of a left ventricular-assisted device was able to return ceramide levels to normal, attenuated PKC activation and improved insulin resistance. This study highlights the clinical relevance of moderate elevations in ceramide content and PKC activation.
Conclusion
In summary, this study provides a novel mechanism by which ceramide may depress cardiac contractility through activation of PKC and subsequent phosphorylation of myofilament proteins and further suggests that accumulation of ceramide in pathological conditions should be considered as a significant factor contributing to the depression in cardiac function.
References
Ayaz-Guner S, Zhang J, Li L, Walker JW, Ge Y (2009) In vivo phosphorylation site mapping in mouse cardiac troponin I by high resolution top-down electron capture dissociation mass spectrometry: Ser22/23 are the only sites basally phosphorylated. Biochemistry 48:8161–8170. doi:10.1021/bi900739f
Bielawska A, Crane HM, Liotta D, Obeid LM, Hannun YA (1993) Selectivity of ceramide-mediated biology. Lack of activity of erythro-dihydroceramide. J Biol Chem 268:26226–26232
Bowman JC, Steinberg SF, Jiang T, Geenen DL, Fishman GI, Buttrick PM (1997) Expression of protein kinase C beta in the heart causes hypertrophy in adult mice and sudden death in neonates. J Clin Invest 100:2189–2195. doi:10.1172/JCI119755
Brenner B (1988) Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc Natl Acad Sci USA 85:3265–3269
Brutsaert DL, Sys SU (1989) Relaxation and diastole of the heart. Physiol Rev 69:1228–1315
Burkart EM, Sumandea MP, Kobayashi T, Nili M, Martin AF, Homsher E, Solaro RJ (2003) Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J Biol Chem 278:11265–11272. doi:10.1074/jbc.M210712200
Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE (2001) A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest 107:813–822. doi:10.1172/JCI10947
Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, Kato T, Khan R, Takayama H, Knoll R, Milting H, Chung CS, Jorde U, Naka Y, Mancini DM, Goldberg IJ, Schulze PC (2012) Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation 125:2844–2853. doi:10.1161/CIRCULATIONAHA.111.060889
Coulton AT, Stelzer JE (2012) Cardiac myosin binding protein C and its phosphorylation regulate multiple steps in the cross-bridge cycle of muscle contraction. Biochemistry 51:3292–3301. doi:10.1021/bi300085x
D’Alessandro ME, Chicco A, Lombardo YB (2008) Dietary fish oil reverses lipotoxicity, altered glucose metabolism, and nPKCepsilon translocation in the heart of dyslipemic insulin-resistant rats. Metabolism 57:911–919. doi:10.1016/j.metabol.2008.02.005
de Tombe PP, Belus A, Piroddi N, Scellini B, Walker JS, Martin AF, Tesi C, Poggesi C (2007) Myofilament calcium sensitivity does not affect cross-bridge activation-relaxation kinetics. Am J Physiol Regul Integr Comp Physiol 292:R1129–R1136. doi:10.1152/ajpregu.00630.2006
Drosatos K, Bharadwaj KG, Lymperopoulos A, Ikeda S, Khan R, Hu Y, Agarwal R, Yu S, Jiang H, Steinberg SF, Blaner WS, Koch WJ, Goldberg IJ (2011) Cardiomyocyte lipids impair beta-adrenergic receptor function via PKC activation. Am J Physiol Endocrinol Metab 300:E489–E499. doi:10.1152/ajpendo.00569.2010
Edman KA, Mattiazzi AR (1981) Effects of fatigue and altered pH on isometric force and velocity of shortening at zero load in frog muscle fibres. J Muscle Res Cell Motil 2:321–334
Ferreira LF, Moylan JS, Gilliam LA, Smith JD, Nikolova-Karakashian M, Reid MB (2010) Sphingomyelinase stimulates oxidant signaling to weaken skeletal muscle and promote fatigue. Am J Physiol Cell Physiol 299:C552–C560. doi:10.1152/ajpcell.00065.2010
Ferreira LF, Moylan JS, Stasko S, Smith JD, Campbell KS, Reid MB (2012) Sphingomyelinase depresses force and calcium sensitivity of the contractile apparatus in mouse diaphragm muscle fibers. J Appl Physiol 112:1538–1545. doi:10.1152/japplphysiol.01269.2011
Finck BN, Han X, Courtois M, Aimond F, Nerbonne JM, Kovacs A, Gross RW, Kelly DP (2003) A critical role for PPARalpha-mediated lipotoxicity in the pathogenesis of diabetic cardiomyopathy: modulation by dietary fat content. Proc Natl Acad Sci USA 100:1226–1231. doi:10.1073/pnas.0336724100
Florea S, Anjak A, Cai WF, Qian J, Vafiadaki E, Figueria S, Haghighi K, Rubinstein J, Lorenz J, Kranias EG (2012) Constitutive phosphorylation of inhibitor-1 at Ser67 and Thr75 depresses calcium cycling in cardiomyocytes and leads to remodeling upon aging. Basic Res Cardiol 107:279. doi:10.1007/s00395-012-0279-z
Gordon AM, Huxley AF, Julian FJ (1966) The variation in isometric tension with sarcomere length in vertebrate muscle fibres. J Physiol 184:170–192
Hannun YA (1996) Functions of ceramide in coordinating cellular responses to stress. Science 274:1855–1859. doi:10.1126/science.274.5294.1855
Hannun YA, Luberto C (2000) Ceramide in the eukaryotic stress response. Trends Cell Biol 10:73–80. doi:10.1016/S0962-8924(99)01694-3
Harrison SM, Bers DM (1990) Modification of temperature dependence of myofilament Ca sensitivity by troponin C replacement. Am J Physiol 258:C282–C288
Hartman TJ, Martin JL, Solaro RJ, Samarel AM, Russell B (2009) CapZ dynamics are altered by endothelin-1 and phenylephrine via PIP2- and PKC-dependent mechanisms. Am J Physiol Cell Physiol 296:C1034–C1039. doi:10.1152/ajpcell.00544.2008
Hinken AC, Hanft LM, Scruggs SB, Sadayappan S, Robbins J, Solaro RJ, McDonald KS (2012) Protein kinase C depresses cardiac myocyte power output and attenuates myofilament responses induced by protein kinase A. J Muscle Res Cell Motil 33:439–448. doi:10.1007/s10974-012-9294-9
Huxley AF (1957) Muscle structure and theories of contraction. Prog Biophys Biophys Chem 7:255–318
Jideama NM, Noland TA Jr, Raynor RL, Blobe GC, Fabbro D, Kazanietz MG, Blumberg PM, Hannun YA, Kuo JF (1996) Phosphorylation specificities of protein kinase C isozymes for bovine cardiac troponin I and troponin T and sites within these proteins and regulation of myofilament properties. J Biol Chem 271:23277–23283
Kirk JA, MacGowan GA, Evans C, Smith SH, Warren CM, Mamidi R, Chandra M, Stewart AF, Solaro RJ, Shroff SG (2009) Left ventricular and myocardial function in mice expressing constitutively pseudophosphorylated cardiac troponin I. Circ Res 105:1232–1239. doi:10.1161/CIRCRESAHA.109.205427
Kobayashi T, Solaro RJ (2005) Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu Rev Physiol 67:39–67. doi:10.1146/annurev.physiol.67.040403.114025
Kooij V, Boontje N, Zaremba R, Jaquet K, dos Remedios C, Stienen GJ, van der Velden J (2010) Protein kinase C alpha and epsilon phosphorylation of troponin and myosin binding protein C reduce Ca2+ sensitivity in human myocardium. Basic Res Cardiol 105:289–300. doi:10.1007/s00395-009-0053-z
Layland J, Grieve DJ, Cave AC, Sparks E, Solaro RJ, Shah AM (2004) Essential role of troponin I in the positive inotropic response to isoprenaline in mouse hearts contracting auxotonically. J Physiol 556:835–847. doi:10.1113/jphysiol.2004.061176
Lee SY, Kim JR, Hu Y, Khan R, Kim SJ, Bharadwaj KG, Davidson MM, Choi CS, Shin KO, Lee YM, Park WJ, Park IS, Jiang XC, Goldberg IJ, Park TS (2012) Cardiomyocyte specific deficiency of serine palmitoyltransferase subunit 2 reduces ceramide but leads to cardiac dysfunction. J Biol Chem 287:18429–18439. doi:10.1074/jbc.M111.296947
Lester JW, Hofmann PA (2000) Role for PKC in the adenosine-induced decrease in shortening velocity of rat ventricular myocytes. Am J Physiol Heart Circ Physiol 279:H2685–H2693
Liu SJ, Kennedy RH (2003) Positive inotropic effect of ceramide in adult ventricular myocytes: mechanisms dissociated from its reduction in Ca2+ influx. Am J Physiol Heart Circ Physiol 285:H735–H744. doi:10.1152/ajpheart.01098.2002
Liu WS, Heckman CA (1998) The sevenfold way of PKC regulation. Cell Signal 10:529–542. doi:10.1016/S0898-6568(98)00012-6
Mohamed AS, Dignam JD, Schlender KK (1998) Cardiac myosin-binding protein C (MyBP-C): identification of protein kinase A and protein kinase C phosphorylation sites. Arch Biochem Biophys 358:313–319. doi:10.1006/abbi.1998.0857
Montgomery DE, Chandra M, Huang Q, Jin J, Solaro RJ (2001) Transgenic incorporation of skeletal TnT into cardiac myofilaments blunts PKC-mediated depression of force. Am J Physiol Heart Circ Physiol 280:H1011–H1018
Noland TA Jr, Guo X, Raynor RL, Jideama NM, Averyhart-Fullard V, Solaro RJ, Kuo JF (1995) Cardiac troponin I mutants. Phosphorylation by protein kinases C and A and regulation of Ca(2+)-stimulated MgATPase of reconstituted actomyosin S-1. J Biol Chem 270:25445–25454
Noland TA Jr, Kuo JF (1991) Protein kinase C phosphorylation of cardiac troponin I or troponin T inhibits Ca2(+)-stimulated actomyosin MgATPase activity. J Biol Chem 266:4974–4978
Noland TA Jr, Kuo JF (1992) Protein kinase C phosphorylation of cardiac troponin T decreases Ca(2+)-dependent actomyosin MgATPase activity and troponin T binding to tropomyosin-F-actin complex. Biochem J 288(Pt 1):123–129
Noland TA Jr, Raynor RL, Jideama NM, Guo X, Kazanietz MG, Blumberg PM, Solaro RJ, Kuo JF (1996) Differential regulation of cardiac actomyosin S-1 MgATPase by protein kinase C isozyme-specific phosphorylation of specific sites in cardiac troponin I and its phosphorylation site mutants. Biochemistry 35:14923–14931. doi:10.1021/bi9616357
Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED, Goldberg IJ (2008) Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res 49:2101–2112. doi:10.1194/jlr.M800147-JLR200
Pellieux C, Montessuit C, Papageorgiou I, Pedrazzini T, Lerch R (2012) Differential effects of high-fat diet on myocardial lipid metabolism in failing and nonfailing hearts with angiotensin II-mediated cardiac remodeling in mice. Am J Physiol Heart Circ Physiol 302:H1795–H1805. doi:10.1152/ajpheart.01023.2011
Puceat M, Clement O, Lechene P, Pelosin JM, Ventura-Clapier R, Vassort G (1990) Neurohormonal control of calcium sensitivity of myofilaments in rat single heart cells. Circ Res 67:517–524
Puglisi JL, Bassani RA, Bassani JW, Amin JN, Bers DM (1996) Temperature and relative contributions of Ca transport systems in cardiac myocyte relaxation. Am J Physiol 270:H1772–H1778
Pyle WG, Chen Y, Hofmann PA (2003) Cardioprotection through a PKC-dependent decrease in myofilament ATPase. Am J Physiol Heart Circ Physiol 285:H1220–H1228. doi:10.1152/ajpheart.00076.2003
Pyle WG, Sumandea MP, Solaro RJ, De Tombe PP (2002) Troponin I serines 43/45 and regulation of cardiac myofilament function. Am J Physiol Heart Circ Physiol 283:H1215–H1224. doi:10.1152/ajpheart.00128.2002
Relling DP, Hintz KK, Ren J (2003) Acute exposure of ceramide enhances cardiac contractile function in isolated ventricular myocytes. Br J Pharmacol 140:1163–1168. doi:10.1038/sj.bjp.0705510
Reuter H, Seuthe K, Korkmaz Y, Gronke S, Hoyer DP, Rottlaender D, Zobel C, Addicks K, Hoyer J, Grimminger P, Brabender J, Wilkie TM, Erdmann E (2012) The G protein Galpha11 is essential for hypertrophic signalling in diabetic myocardium. Int J Cardiol. doi:10.1016/j.ijcard.2012.04.039
Sadayappan S, Gulick J, Osinska H, Barefield D, Cuello F, Avkiran M, Lasko VM, Lorenz JN, Maillet M, Martin JL, Brown JH, Bers DM, Molkentin JD, James J, Robbins J (2011) A critical function for Ser-282 in cardiac Myosin binding protein-C phosphorylation and cardiac function. Circ Res 109:141–150. doi:10.1161/CIRCRESAHA.111.242560
Sadayappan S, Gulick J, Osinska H, Martin LA, Hahn HS, Dorn GW 2nd, Klevitsky R, Seidman CE, Seidman JG, Robbins J (2005) Cardiac myosin-binding protein-C phosphorylation and cardiac function. Circ Res 97:1156–1163. doi:10.1161/01.RES.0000190605.79013.4d
Sancho Solis R, Ge Y, Walker JW (2008) Single amino acid sequence polymorphisms in rat cardiac troponin revealed by top-down tandem mass spectrometry. J Muscle Res Cell Motil 29:203–212. doi:10.1007/s10974-009-9168-y
Scruggs SB, Walker LA, Lyu T, Geenen DL, Solaro RJ, Buttrick PM, Goldspink PH (2006) Partial replacement of cardiac troponin I with a non-phosphorylatable mutant at serines 43/45 attenuates the contractile dysfunction associated with PKCepsilon phosphorylation. J Mol Cell Cardiol 40:465–473. doi:10.1016/j.yjmcc.2005.12.009
Shattock MJ, Bers DM (1987) Inotropic response to hypothermia and the temperature-dependence of ryanodine action in isolated rabbit and rat ventricular muscle: implications for excitation-contraction coupling. Circ Res 61:761–771
Sheehan KA, Arteaga GM, Hinken AC, Dias FA, Ribeiro C, Wieczorek DF, Solaro RJ, Wolska BM (2011) Functional effects of a tropomyosin mutation linked to FHC contribute to maladaptation during acidosis. J Mol Cell Cardiol 50:442–450. doi:10.1016/j.yjmcc.2010.10.032
Solaro RJ (2008) Multiplex kinase signaling modifies cardiac function at the level of sarcomeric proteins. J Biol Chem 283:26829–26833. doi:10.1074/jbc.R800037200
Strang KT, Moss RL (1995) Alpha 1-adrenergic receptor stimulation decreases maximum shortening velocity of skinned single ventricular myocytes from rats. Circ Res 77:114–120
Sumandea MP, Pyle WG, Kobayashi T, de Tombe PP, Solaro RJ (2003) Identification of a functionally critical protein kinase C phosphorylation residue of cardiac troponin T. J Biol Chem 278:35135–35144. doi:10.1074/jbc.M306325200
Takeishi Y, Ping P, Bolli R, Kirkpatrick DL, Hoit BD, Walsh RA (2000) Transgenic overexpression of constitutively active protein kinase C epsilon causes concentric cardiac hypertrophy. Circ Res 86:1218–1223
Tong CW, Stelzer JE, Greaser ML, Powers PA, Moss RL (2008) Acceleration of crossbridge kinetics by protein kinase A phosphorylation of cardiac myosin binding protein C modulates cardiac function. Circ Res 103:974–982. doi:10.1161/CIRCRESAHA.108.177683
Warren CM, Arteaga GM, Rajan S, Ahmed RP, Wieczorek DF, Solaro RJ (2008) Use of 2-D DIGE analysis reveals altered phosphorylation in a tropomyosin mutant (Glu54Lys) linked to dilated cardiomyopathy. Proteomics 8:100–105. doi:10.1002/pmic.200700772
Watts JD, Gu M, Patterson SD, Aebersold R, Polverino AJ (1999) On the complexities of ceramide changes in cells undergoing apoptosis: lack of evidence for a second messenger function in apoptotic induction. Cell Death Differ 6:105–114. doi:10.1038/sj.cdd.4400472
Weith A, Sadayappan S, Gulick J, Previs MJ, Vanburen P, Robbins J, Warshaw DM (2012) Unique single molecule binding of cardiac myosin binding protein-C to actin and phosphorylation-dependent inhibition of actomyosin motility requires 17 amino acids of the motif domain. J Mol Cell Cardiol 52:219–227. doi:10.1016/j.yjmcc.2011.09.019
Wolska BM, Kitada Y, Palmiter KA, Westfall MV, Johnson MD, Solaro RJ (1996) CGP-48506 increases contractility of ventricular myocytes and myofilaments by effects on actin-myosin reaction. Am J Physiol 270:H24–H32
Wu G, Toyokawa T, Hahn H, Dorn GW 2nd (2000) Epsilon protein kinase C in pathological myocardial hypertrophy. Analysis by combined transgenic expression of translocation modifiers and Galphaq. J Biol Chem 275:29927–29930. doi:10.1074/jbc.C000380200
Young LH, Balin BJ, Weis MT (2005) Go 6983: a fast acting protein kinase C inhibitor that attenuates myocardial ischemia/reperfusion injury. Cardiovasc Drug Rev 23:255–272. doi:10.1111/j.1527-3466.2005.tb00170.x
Young ME, Guthrie PH, Razeghi P, Leighton B, Abbasi S, Patil S, Youker KA, Taegtmeyer H (2002) Impaired long-chain fatty acid oxidation and contractile dysfunction in the obese Zucker rat heart. Diabetes 51:2587–2595
Zhang J, Dong X, Hacker TA, Ge Y (2010) Deciphering modifications in swine cardiac troponin I by top-down high-resolution tandem mass spectrometry. J Am Soc Mass Spectrom 21:940–948. doi:10.1016/j.jasms.2010.02.005
Zhou YT, Grayburn P, Karim A, Shimabukuro M, Higa M, Baetens D, Orci L, Unger RH (2000) Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci USA 97:1784–1789. doi:10.1073/pnas.97.4.1784
Acknowledgments
The authors would like to acknowledge Dr. Suresh Govidan and Dr. Xiang Ji for their technical assistance with the cMyBP-C immunoblots. This research was supported by an American Heart Association, Midwest Affiliate pre-doctoral fellowship to JNS, NIH research grants R01 HL-64035 (RJS and BMW), R01 HL-105826 and K02 HL-114749 (SS), RO1 HL-081680 (DFW), and PO1 HL-62426 (Project 1 and Core C to RJS and CMW).
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Simon, J.N., Chowdhury, S.A.K., Warren, C.M. et al. Ceramide-mediated depression in cardiomyocyte contractility through PKC activation and modulation of myofilament protein phosphorylation. Basic Res Cardiol 109, 445 (2014). https://doi.org/10.1007/s00395-014-0445-6
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-014-0445-6