
Abstract
Protein kinase G type I (PKGI) plays a critical role in survival signaling of pre- and postconditioning downstream of cardiac cGMP. However, it is unclear whether PKGI exerts its protective effects in the cardiomyocyte or if other cardiac cell types are involved, and whether nitric oxide (NO) metabolism can target cardiomyocyte mitochondria independently of cGMP/PKGI. We tested whether protection against reperfusion injury by ischemic postconditioning (IPost), soluble guanylyl cyclase (sGC) activation and inhibition, adenosine A2B receptor (A2BAR) agonist, phosphodiesterase type-5 (PDE-5) inhibitor, or mitochondria-targeted S-nitrosothiol (MitoSNO) was affected by a cardiomyocyte-specific ablation of the PKGI gene in the mouse (CMG-KO). In situ hearts underwent 30 min of regional ischemia followed by 2 h of reperfusion. As expected, in CMG-CTRs all interventions at early reperfusion lead to profound infarct size reduction: IPost (six cycles of 10-s reperfusion and 10-s coronary occlusion) with or without treatment with the sGC inhibitor ODQ, treatment with the specific sGC activator BAY58-2667 (BAY58), the selective A2BAR agonist BAY60-6583 (BAY60), PDE-5 inhibitor sildenafil, and MitoSNO. MitoSNO accumulates within mitochondria, driven by the membrane potential, where it generates NO· and S-nitrosates thiol proteins. In contrast, the hearts of CMG-KO animals were not protected by BAY58 and sildenafil, whereas the protective effects of IPost, IPost with ODQ, BAY60, and MitoSNO were unaffected by the lack of PKGI. Taken together, PKGI is important for the protection against ischemia reperfusion injury afforded by sGC activation or PDE-5 inhibition. However, the beneficial effects of IPost, activation of the A2BAR, as well as the direct effects via mitochondrial S-nitrosation do not depend on PKGI in cardiomyocytes.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The first minutes of reperfusion represent a window of opportunity for triggering signaling pathways which ultimately lead to protection against reperfusion injury in the heart. While the cardiomyocyte is the critical cell type to be protected, it is not clear whether the protective signaling occurs exclusively within the myocyte itself or whether signals derived from other cardiac cell types, such as endothelial cells or smooth muscle cells, contribute to cardiomyocyte survival. In many cell types increased concentrations of cyclic guanosine-3′, 5′-monophosphate (cGMP) activate protein kinase G (PKG). In addition, cGMP is expected to have many effects other than PKG activation since cGMP binds to, and modulates multiple effectors within a single cell [4]. The two PKG genes, prkg1 and prkg2, encode three different enzymes, PKGIα/β and PKGII, respectively, with the PKGIα isozyme being predominantly expressed in the heart [11]. Pharmacological agents that elevate the myocardial cGMP content and therefore should activate PKGI have been shown to be beneficial for the heart upon ischemia/reperfusion injury [5, 8, 16], presumably via cGMP/PKGI-dependent transmission of a yet unknown protective signal from the cytosol to the mitochondria [3]. Importantly, it was demonstrated that the infarct size was reduced when cGMP elevating drugs were administered at the onset of reperfusion [2]. Most of these studies have been performed in models assessing infarct size either in isolated or in situ hearts, without distinguishing between signaling in cardiomyocytes or other cardiac cell types. In addition, the myocardial cGMP content and the availability of components within the heart that respond to modulators of the cGMP pathway may change upon ischemia. In a recent report it was shown that the absence of PKGI in the cardiomyocyte did not affect the hypertrophic growth response induced by either isoproterenol or transverse aortic constriction [17], suggesting that the cardiomyocyte might not be the primary location for protective cGMP/PKGI signaling. Furthermore, Sun et al. [23] used pharmacological inhibitors to show that ischemic preconditioning is unaffected by blocking the sGC/cGMP/PKG pathway. In the present study we used the Cre/loxP system to inactivate endogenous PKGI selectively in the cardiomyocyte. We show that the selective inactivation of PKGI in cardiomyocytes does not affect the protective properties of ischemic postconditioning, adenosine receptor stimulation, and NO acting via S-nitrosation of mitochondrial proteins, whereas the protective signaling via sGC activation and phosphodiesterase type-5 inhibition was lost in the absence of PKGI. These results show that sGC/cGMP-dependent protection against reperfusion injury signaling requires PKGI in the cardiomyocytes.
Methods
An expanded Methods section is available in the Online Supplement.
Mice
Conditional mice with floxed PKGI alleles (L2) were described earlier [25]. To generate cardiomyocyte-specific PKGI knockout animals mice with floxed PKGI alleles (genotype: PKGIL2/L2) were crossed to the MLC2α-Cre transgenic mouse line (genotype: MLC2α-Cretg/+; PKGI+/L-). The Cre-mediated recombination of the L2 allele produced the PKGI knockout (L-) specifically in atrial and ventricular cardiomyocytes but not in other tissues. For experiments cardiomyocyte-specific PKGI knockout mice (CMG-KO; genotype: MLC2α-Cretg/+; PKGIL2/L-) were compared with either control mice (CMG-CTR; MLC2α-Cretg/+; PKGI+/L2) from the same litters on a C57BL/6 genetic background, or with C57BL/6 itself.
Open-chest mouse model
An open-chest, in situ mouse heart model was used as recently described [18]. Mice were anesthetized with pentobarbital sodium (70 mg/kg i.p.), adequacy of anesthesia was monitored via corneal and withdraw reflexes, and additional anesthesia was administered as needed throughout the experiment. All hearts underwent 30 min of coronary artery occlusion followed by 2 h of reperfusion. IPost was effected with six cycles of 10-s reperfusion and 10-s coronary artery occlusion following the index ischemia. BAY60 (10 μg/kg) was applied intraatrially as two separate boluses, 5 min before and 15 min after the onset of reperfusion. BAY58 (53.6 μg/kg) was given as an intravenous bolus 5 min before the onset of reperfusion followed by an infusion (1.25 μg/kg/min) for 65 min. MitoSNO (100 ng/kg), as well as sildenafil (2 μg/kg) was administered as one bolus 5 min before the onset of reperfusion into the left atrium. 1H-[1, 2, 4] oxadiazolo[4,3-a]quinoxal in-1-one (ODQ, 2 mg/kg) was given intraperitoneally 20 min before the onset of reperfusion. For all protective interventions, we chose drug applications which have been previously reported to be highly effective.
Statistics
All values are presented as mean ± SEM. The significance of differences between groups was determined by one-way ANOVA with Fisher’s LDS post hoc test. A value of p < 0.05 was considered significant.
Results
Infarct size measurements
Figure 1 depicts the results for CMG-CTR mice, which are C57BL/6-derived offspring from the same breeding stock used to produce the cardiomyocyte-specific PKGI knockouts (CMG-KO). As expected, infarct size after 30-min LAD occlusion and 2-h reperfusion showed significant cardioprotection with either ischemic postconditioning (IPost), the A2BAR agonist BAY60, the S-nitrosating compound MitoSNO, sGC activation with BAY58, or the PDE-5 inhibitor sildenafil. Interestingly, the selective sGC inhibitor ODQ could not block the IPost’s protective effects. The results obtained in the CMG-CTR mice (Fig. 1) agree well with previous reports using identical interventions in C57BL/6 wild-type mice, ruling out any unexpected independent effects of the genetic background (Table 1).

Infarct size in respective control mice of cardiomyocyte-specific PKGI knockouts (CMG-CTR). All interventions applied at reperfusion resulted in a profound decrease of infarct size compared with control in CMG-CTR: ischemic postconditioning (IPost), the activation of the A2BAR subtype with BAY60-6583 (BAY60), S-nitrosylation of mitochondrial proteins via MitoSNO, sGC activation with BAY58-2667 (BAY58), or PDE-5 inhibition with sildenafil. IPost’s protection could not be blocked by the sGC inhibitor ODQ. Infarction was measured by double staining with Evan’s Blue and TTC and is expressed as percentage of the area at risk (AAR). Open symbols represent individual experiments while closed symbols are the mean ± SEM. *p < 0.05 vs. control
Figure 2 summarizes the results obtained in cardiomyocyte-specific PKGI knockout mice (CMG-KO). Hearts from these mice could still be postconditioned and protected by BAY60 and MitoSNO, but activating the NO/PKG pathway with the sGC activator BAY58 or sildenafil failed to afford cardioprotection. Protection via postconditioning was still present when sGC was inhibited with ODQ in these mice lacking cardiomyocyte PKGI.

Infarct size in cardiomyocyte-specific PKGI knockout mice (CMG-KO). aWhereas BAY58 and sildenafil failed to protect CMG-KOs, IPost, BAY60, MitoSNO, and IPost in the presence or absence of ODQ still afforded protection. Open symbols represent individual experiments while closed symbols are the mean ± SEM. b Representative images of infarcts from the CMG-KO experiments (blue indicates retrograde Evan’s Blue staining; red and white, AAR; white; infarcted tissue). *p < 0.05 vs. control, BAY58, and sildenafil
The results from the infarct size measurements are mirrored by the amount of serum troponin I found at the end of the reperfusion phase (Fig. 3).

Plasma levels of cardiac troponin I (cTnI) at the end of reperfusion. Plasma level of cTnI at the end of open-chest mouse experiments in a control (CMG-CTR) and b cardiomyocyte-specific PKGI knockout animals (CMG-KO).BAY60: BAY60-6583, A2BAR agonist; BAY58: BAY58-2667, sGC activator; IPost: ischemic postconditioning; MitoSNO: mitochondria-selective S-nitrosating agent; ODQ: selective sGC inhibitor; sildenafil: PDE-5 blocker. *p < 0.05 vs. control (a), and p < 0.05 vs. control, BAY58, and sildenafil (b)
PKG protein expression
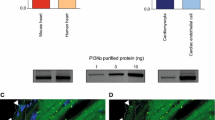
Western blot analysis and immunohistochemistry were performed to demonstrate that ablation of PKGI was cardiomyocyte-specific (Fig. 4). PKG protein is absent in CMG-KO mice in the ventricle (Fig. 4a) but is still present in the cerebellum (Fig 4b). By immunohistochemistry PKGI protein was detected in the myocardium of heart sections from control mice but was absent from heart of CMG-KO animals (Fig. 4c). Control experiments in which the same protocol was applied to sections but without the primary antibody showed no staining.

Cardiomyocyte-specific ablation of PKGI in CMG-KO hearts. Western Blot analysis of protein homogenates from a the ventricle and b cerebellum of CMG-CTR and CMG-KO animals. By using polyclonal antibodies that specifically detect either the PKGIα isoform or both PKGI isoforms (PKGcom) a significant reduction in the PKGI protein content was detectable in lysates obtained from CMG-KO hearts. Antibodies detecting GAPDH and the cardiac isoform of α-actinin were used to identify equal loading of the gels. c By immunohistochemistry the PKGI protein was detected in the myocardium of heart sections from CMG-CTR mice (upper left panel), but was close to absent from heart muscles of CMG-KO animals (upper right panel). No staining was noted when the same staining protocol was applied to the sections when the primary antibodies were omitted (lower panels) (scale bars, 50 μm)
Discussion
It has been demonstrated that activation of PKGI at the onset of reperfusion leads to a profound reduction of infarct size in various models and species [2, 6, 14, 19]. We recently demonstrated that both, IPost and the selective A2BAR agonist BAY60, were able to reduce infarct size in wild-type C57BL/6 mice [18], and in the present study, we found the sGC activator BAY58 and PDE-5 inhibitor sildenafil to be equally protective in the CMG-CTR mice, which were offspring of the same breeding stock used to produce the cardiomyocyte-specific PKGI knockouts (CMG-KOs). These results agree with previous reports using either IPost, sGC activation, PDE-5 inhibition, or an A2BAR agonist to protect hearts in various models and species [13, 15, 18, 21]. In addition, we could confirm in our postconditioning model the cardioprotection by S-nitrosation of proteins that had previously been reported for preconditioning [23]. Here we used the novel S-nitrosating compound MitoSNO, which comprises the NO donor SNAP conjugated to a triphenylphosphonium (TPP) moiety. The lipophilic TPP allows MitoSNO to pass rapidly through membranes driven by the membrane potential and therefore accumulate several-hundred-fold within the mitochondria [12]. It has been shown that MitoSNO is highly selective at S-nitrosating mitochondrial proteins [20]. There is considerable evidence that mitochondria are the decisive organelle that has to be protected during I/R injury [1, 9, 10].
Interestingly, when we used mice lacking PKGI selectively in cardiomyocytes (CMG-KO), the hearts could still be postconditioned even in the presence of a selective, widely used sGC inhibitor. In addition, MitoSNO, as well as the highly selective A2B AR agonist BAY60, were still protective when applied at reperfusion in these CMG-KO animals. On the other hand, the selective sGC activator BAY58 and sildenafil failed to protect. While IPost signaling seems to bypass PKGI, and adenosine receptor stimulation could be downstream, these findings support previous reports that PKGI within the cardiomyocyte is indeed an important effector of the cGMP pathway, which counteracts cardiac damage.
Recently, it has been shown that the genetic ablation of PKGI in the cardiomyocyte had no effect on changes in the heart following various hypertrophic stimuli [17]. While in the earlier study mice were analyzed that expressed PKGI in all smooth muscles including the vasculature of the heart on a PKGI-negative background (PKGI “rescue mice”), we used knockout animals where PKGI is inactivated specifically in the heart muscle but was still present in all other organs and cells.
Using knockout mice lacking the functionally predominant isoform of sGC in heart (sGCα1), it has been shown that sGCα1 is not required for cardioprotection mediated by ischemic postconditioning [22]. It was suggested that the second isoform, sGCα2, may provide sufficient activity for inducing cardioprotective signaling, since ODQ, which inhibits all sGC isoforms, blocked the protection of ischemic preconditioning in sGCα1 knockout mice. With the mice used in this study, we cannot rule out a similar compensation mechanism with the action of other PKG isoforms, but there are various reports identifying PKGI as the predominant, if not the only, PKG isoform in the cardiomyocyte [26]. In addition, in our hands ODQ could not block the protective effect of postconditioning further suggesting a PKG-independent way to protect. Our data agree with a recent report from Elisabeth Murphy’s group which showed that ODQ failed to inhibit the cardioprotective effect of ischemic preconditioning in isolated Langendorff-perfused hearts [23].
We found that the highly selective A2BAR agonist BAY60 still reduced infarct size in hearts lacking PKGI (CMG-KO) hearts. This finding is supported by Kuno et al. [15] who showed that the A2BAR most likely acts downstream of PKGI, since selective A2BAR inhibitors abolished the beneficial effects of PKG on isolated rabbit hearts. In addition, we recently suggested an intracellular location of the A2BAR at mitochondria, which is consistent with A2BAR acting downstream of the PKG signaling [7].
The selective mitochondrial nitrosating agent MitoSNO proved to be equally protective in CMG-KOs, again indicating that the protection conferred by mitochondrial S-nitrosation is not dependent on PKGI in the cardiomyocyte. There is growing evidence that protein nitrosation is cardioprotective, but little is known about the underlying mechanism(s) [24]. Furthermore, it cannot be ruled out that MitoSNO acts to protect the heart from reperfusion injury in a manner that depends on PKGI in other cardiac cell types or other models. Future studies that make use of, e.g., platelet, smooth muscle, and/or endothelial cell specific PKGI knockouts are needed to eliminate this possibility.
Taken together, we present compelling data supporting the importance of cardiomyocyte PKGI for PKGI-dependent protection against ischemia/reperfusion injury, while IPost, A2BAR activation and direct effects of NO via S-nitrosation of mitochondrial proteins seem to afford protection either by bypassing PKGI or by acting independently or downstream of it (Fig. 5). Using this conditional knockout approach to inactivate PKGI we were able to differentiate between cGMP/PKGI signaling in myocytes and other cardiac cell types during IPost’s protection in vivo. Further studies are needed to fully explore the signaling pathways involved as well as the possible importance of the non-myocyte cell populations and different PKG isoforms in cardioprotection.

Hypothetical model for protective signaling. Nitric oxide (NO) can cause protection against ischemia/reperfusion injury through prevention of mitochondrial permeability transition (mPTP) via sGC, cGMP, and PKG. This pathway can be bypassed and cause NO-dependent protection through direct S-nitrosation of mitochondrial proteins (X-SNO). It is not clear how A2B adenosine receptors (A2BAR) fit into the scheme, but they seem to be either downstream or parallel of the “classical” NO/PKG pathway leading to prevention of mPTP
References
Boengler K, Hilfiker-Kleiner D, Heusch G, Schulz R (2010) Inhibition of permeability transition pore opening by mitochondrial STAT3 and its role in myocardial ischemia/reperfusion. Basic Res Cardiol 105:771–785. doi:10.1007/s00395-010-0124-1
Burley DS, Ferdinandy P, Baxter GF (2007) Cyclic GMP and protein kinase-G in myocardial ischaemia-reperfusion: opportunities and obstacles for survival signaling. Br J Pharmacol 152:855–869. doi:10.1038/sj.bjp.0707409
Costa AD, Garlid KD, West IC, Lincoln TM, Downey JM, Cohen MV, Critz SD (2005) Protein kinase G transmits the cardioprotective signal from cytosol to mitochondria. Circ Res 97:329–336. doi:10.1161/01.RES.0000178451.08719.5b
Francis SH, Busch JL, Corbin JD, Sibley D (2010) cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol Rev 62:525–563. doi:10.1124/pr.110.002907
Garcia-Dorado D, Agulló L, Sartorio CL, Ruiz-Meana M (2009) Myocardial protection against reperfusion injury: the cGMP pathway. Thromb Haemost 101:635–642. doi:10.1160/TH08-11-0764
Gorbe A, Giricz Z, Szunyog A, Csont T, Burley DS, Baxter GF, Ferdinandy P (2010) Role of cGMP-PKG signaling in the protection of neonatal rat cardiac myocytes subjected to simulated ischemia/reoxygenation. Basic Res Cardiol 105:643–650. doi:10.1007/s00395-010-0097-0
Grube K, Rüdebusch J, Xu Z, Böckenholt T, Methner C, Müller T, Cuello F, Zimmermann K, Yang X, Felix SB, Cohen MV, Downey JM, Krieg T (2011) Evidence for an intracellular localization of the adenosine A2B receptor in rat cardiomyocytes. Basic Res Cardiol 106:385–396. doi:10.1007/s00395-011-0151-6
Heusch G, Boengler K, Schulz R (2008) Cardioprotection: nitric oxide, protein kinases, and mitochondria. Circulation 118:1915–1919. doi:10.1161/CIRCULATIONAHA.108.805242
Heusch G, Boengler K, Schulz R (2010) Inhibition of mitochondrial permeability transition pore opening: the Holy Grail of cardioprotection. Basic Res Cardiol 105:151–154. doi:10.1007/s00395-009-0080-9
Heusch G, Musiolik J, Gedik N, Skyschally A (2011) Mitochondrial STAT3 activation and cardioprotection by ischemic postconditioning in pigs with regional myocardial ischemia/reperfusion. Circ Res 109:1302–1308. doi:10.1161/CIRCRESAHA.111.255604
Hofmann F, Bernhard D, Lukowski R, Weinmeister P (2009) cGMP regulated protein kinases (cGK). Handb Exp Pharmacol 191:137–162. doi:10.1007/978-3-540-68964-5_8
James AM, Sharpley MS, Manas AR, Frerman FE, Hirst J, Smith RA, Murphy MP (2007) Interaction of the mitochondria-targeted antioxidant MitoQ with phospholipid bilayers and ubiquinone oxidoreductases. J Biol Chem 282:14708–14718. doi:10.1074/jbc.M611463200
Korkmaz S, Radovits T, Barnucz E, Hirschberg K, Neugebauer P, Loganathan S, Veres G, Páli S, Seidel B, Zöllner S, Karck M, Szabó G (2009) Pharmacological activation of soluble guanylate cyclase protects the heart against ischemic injury. Circulation 120:677–686. doi:10.1161/CIRCULATIONAHA.109.870774
Krieg T, Liu Y, Rütz T, Methner C, Yang XM, Dost T, Felix SB, Stasch JP, Cohen MV, Downey JM (2009) BAY 58–2667, a nitric oxide-independent guanylyl cyclase activator, pharmacologically post-conditions rabbit and rat hearts. Eur Heart J 30:1607–1613. doi:10.1093/eurheartj/ehp143
Kuno A, Solenkova NV, Solodushko V, Dost T, Liu Y, Yang XM, Cohen MV, Downey JM (2008) Infarct limitation by a protein kinase G activator at reperfusion in rabbit hearts is dependent on sensitizing the heart to A2b agonists by protein kinase C. Am J Physiol Heart Circ Physiol 295:H1288–H1295. doi:10.1152/ajpheart.00209.2008
Li Q, Guo Y, Wu W-J, Ou Q, Zhu X, Tan W, Yuan F, Chen N, Dawn B, Luo L, O’Brian E, Bolli R (2011) Gene transfer as a strategy to achieve permanent cardioprotection I: rAAV-mediated gene therapy with inducible nitric oxide synthase limits infarct size 1 year later without adverse functional consequences. Basic Res Cardiol 106:1355–1366. doi:10.1007/s00395-011-0207-7
Lukowski R, Rybalkin SD, Loga F, Leiss V, Beavo JA, Hofmann F (2010) Cardiac hypertrophy is not amplified by deletion of cGMP-dependent protein kinase I in cardiomyocytes. Proc Natl Acad Sci USA 107:5646–5651. doi:10.1073/pnas.1001360107
Methner C, Schmidt K, Cohen MV, Downey JM, Krieg T (2010) Both A2a and A2b adenosine receptors at reperfusion are necessary to reduce infarct size in mouse hearts. Am J Physiol Heart Circ Physiol 299:H1262–H1264. doi:10.1152/ajpheart.00181.2010
Penna C, Cappello S, Mancardi D, Raimondo S, Rastaldo R, Gattullo D, Losano G, Pagliaro P (2006) Post-conditioning reduces infarct size in the isolated rat heart: role of coronary flow and pressure and the nitric oxide/cGMP pathway. Basic Res Cardiol 101:168–179. doi:10.1007/s00395-005-0543-6
Prime TA, Blaikie FH, Evans C, Nadtochiy SM, James AM, Dahm CC, Vitturi DA, Patel RP, Hiley CR, Abakumova I, Requejo R, Chouchani ET, Hurd TR, Garvey JF, Taylor CT, Brookes PS, Smith RA, Murphy MP (2009) A mitochondria-targeted S-nitrosothiol modulates respiration, nitrosates thiols, and protects against ischemia-reperfusion injury. Proc Natl Acad Sci USA 106:10764–10769. doi:10.1073/pnas.0903250106
Salloum FN, Abbate A, Das A, Houser JE, Mudrick CA, Qureshi IZ, Hoke NN, Roy SK, Brown WR, Prabhakar S, Kukreja RC (2008) Sildenafil (Viagra) attenuates ischemic cardiomyopathy and improves left ventricular function in mice. Am J Physiol Heart Circ Physiol 294:H1398–H1406. doi:10.1152/ajpheart.91438.2007
Sips PY, Brouckaert P, Ichinose F (2011) The alpha1 isoform of soluble guanylate cyclase regulates cardiac contractility but is not required for ischemic preconditioning. Basic Res Cardiol 106:635–643. doi:10.1007/s00395-011-0167-y
Sun J, Aponte AM, Kohr MJ, Tong G, Steenbergen C, Murphy E (2012) Essential role of nitric oxide in acute ischemic preconditioning: S-Nitros(yl)ation versus sGC/cGMP/PKG signaling? Free Radic Biol Med 54:105–112. doi:10.1016/j.freeradbiomed.2012.09.005
Sun J, Murphy E (2010) Protein S-nitrosylation and cardioprotection. Circ Res 106:285–296. doi:10.1161/CIRCRESAHA.109.209452
Wegener JW, Nawrath H, Wolfsgruber W, Kühbandner S, Werner C, Hofmann F, Feil R (2002) cGMP-dependent protein kinase I mediates the negative inotropic effect of cGMP in the murine myocardium. Circ Res 90:18–20. doi:10.1161/hh0102.103222
Wollert KC, Fiedler B, Gambaryan S, Smolenski A, Heineke J, Butt E, Trautwein C, Lohmann SM, Drexler H (2002) Gene transfer of cGMP-dependent protein kinase I enhances the antihypertrophic effects of nitric oxide in cardiomyocytes. Hypertension 39:87–92. doi:10.1161/hy1201.097292
Acknowledgments
We thank Ines Suhr, Institute of Clinical Chemistry, University of Greifswald, for part of the troponin analysis. BAY60 and BAY58 were kindly provided by Bayer Healthcare, Wuppertal, Germany. The study was supported by grants from the Deutsche Forschungsgemeinschaft (T.K., R.L., and F.H), the Academic Research Collaboration Program of the British Council and the German Academic Exchange Service (T.K. and K.G.), the Isaac Newton Trust, Trinity College, Cambridge (T.K. and C.M.), and the Eliteprogramm für Postdocs of the Baden-WürttembergStiftung (R.L.).
Conflict of interest
None declared.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Methner, C., Lukowski, R., Grube, K. et al. Protection through postconditioning or a mitochondria-targeted S-nitrosothiol is unaffected by cardiomyocyte-selective ablation of protein kinase G. Basic Res Cardiol 108, 337 (2013). https://doi.org/10.1007/s00395-013-0337-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00395-013-0337-1