
Abstract
We examined the impact of acute myocardial ischemia followed by reperfusion (AMI-R) on local and circulating renin–angiotensin system (RAS) in a swine model. The mid left anterior descending artery (n = 6) was occluded for 1 h, followed by reperfusion for 2 h. Monastryl blue/triphenyl tetrazolium chloride staining identified the area-at-risk (AAR) and infarction. A second group of control animals underwent sham operations (C: n = 4). Myocardial expression of angiotensinogen (AGT), renin, chymase, angiotensin converting enzyme (ACE), angiotensin II (Ang II), Ang II type1 receptor (AT1R) and Ang II type 2 receptor (AT2R) in the AAR and the non-ischemic left ventricle (NLV) was assessed. Serum level of these proteins at baseline and at the end of reperfusion was also examined. Chymase (P < 0.05), ACE (P < 0.05), Ang II (P < 0.05), AT1R (P < 0.05) and AT2R (P < 0.05) expressions were found to be significantly higher in the AAR compared to the NLV and C whereas no significant differences were found for AGT (P = 0.58) and renin (P = 0.38). Serum concentration of ACE was significantly higher at the end of reperfusion than at baseline (P < 0.01), whereas no significant difference was found for chymase (P = 0.71), AGT (P = 0.57) and Ang II (P = 0.19). Immunohistochemistry of myocardial sections demonstrated significantly higher expression of ACE (P = 0.02), AT1R (P = 0.01), AT2R (P = 0.02) and Ang II (P < 0.01) in the AAR as compared to the NLV, whereas no significant difference was found for renin (P = 0.39). In conclusion, AMI-R resulted in significantly higher expression of specific cardiac RAS components in AAR compared to the NLV in the acute period.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The renin–angiotensin system (RAS) is a well-known regulator of blood pressure, tissue perfusion and organ functions. Recently, the RAS has been implicated in conditions such as atrial fibrillation, and has been shown to interact with the bradykinin, prostaglandins, and aldosterone pathways [19, 46, 59]. In pathological conditions, RAS is an important modulator of myocardial remodeling. The RAS modulates myocardial and vascular injury after ischemia. Angiotensin converting enzyme (ACE) inhibition can reduce chronic myocardial remodeling following acute myocardial infarction in part by reducing myocardial afterload and increasing collateral flow [15], and it also directly affects specific cellular pathways that are involved in myocardial remodeling [58].
The RAS exists in two distinct systems in multiple organs: circulating and local. Each system demonstrates independent regulation and pathophysiologic attributes [27–29]. The circulatory RAS involves angiotensin II (Ang II) synthesized in the circulation, whereas in the local RAS, Ang II is synthesized in the tissue. Recently, it has been reported that expression of chymase, which is an enzyme responsible for Ang II generation in the heart [29, 56], and angiotensin-converting enzyme (ACE) is increased during acute myocardial infarction in the rat or hamster model [21, 34]. However, there have been numerous studies to investigate the relationship between myocardial ischemia and the RAS [11, 14–16, 18, 20, 21, 34, 40, 58], none have examined the effects of the local RAS in the heart after acute myocardial ischemia followed by reperfusion (AMI-R) in clinically relevant large animal models. Previous clinical studies are limited with regard to presenting mechanistic information requiring invasive measures such as regional myocardial function or molecular studies. In this study, we used a swine model of AMI-R to examine the expression of the RAS components in both myocardial tissue and blood serum.
Methods
Animals were housed individually and provided with laboratory chow and water ad libitum. All experiments were approved by the Beth Israel Deaconess Medical Center animal care and use committee and the Harvard Medical Area standing committee on animals. The experiments conformed to the US National Institutes of Health guidelines regulating the care and use of laboratory animals (NIH publication 5377-3, 1996).
Experimental design
Six male Yucatan swine (23.3 ± 2.3 kg) were used. Animals were subjected to regional left ventricular (LV) ischemia by left anterior descending (LAD) arterial occlusion distal to the second diagonal branch for 60 min. Following ischemia, the myocardium was reperfused for 120 min. Arterial blood gas (ABG), arterial blood pressure, hematocrit (Hct), LV pressure, heart rate (HR), EKG, O2 saturation and core body temperature were measured and recorded. Myocardial segmental shortening in the long-axis (parallel to the LAD) and short-axis (perpendicular to the LAD) were recorded at baseline prior to the onset of the ischemia, and prior to harvest. At the completion of the protocol, the heart was excised and tissue samples from ischemic-reperfused and non-ischemic left ventricle were collected for analysis as described below. Thus, each pig served as its own control with regard to molecular studies. Four additional control animals (19.1 ± 3.1 kg) underwent sham operations, which did not include ligation of the LAD. Clinical and physiological functional measurement and cardiac harvest were carried out in the same manner described above.
Surgical protocol
Swine were sedated with ketamine hydrochloride (20 mg/kg, intramuscularly, Abbott Laboratories, North Chicago, IL, USA) and anesthetized with a bolus infusion of thiopental sodium (Baxter Healthcare Corporation, Inc., Deerfield, IL; 5.0–7.0 mg/kg intravenously), followed by endotracheal intubation. Ventilation with a volume-cycled ventilator (model Narkomed II-A; North American Drager, Telford, PA, USA; oxygen, 40%; tidal volume, 12.5 ml/kg; ventilation rate, 9–10 breaths/min; positive end-expiratory pressure, 3 cm H2O; inspiratory to expiratory time, 1:2) was utilized. General endotracheal anesthesia was established with 3.0% isoflurane (Ultane; Abbott Laboratories) at the beginning of the surgical preparation, and maintained with 1.0% throughout the experiment. One liter of Lactated Ringer’s intravenous (IV) fluid was administered after induction of anesthesia and continued throughout the surgical protocol at 150 ml/h. A right groin dissection was performed and the femoral vein and common femoral artery were isolated and cannulated utilizing eight French sheaths (Cordis Corporation, Miami, FL, USA). The right femoral vein was cannulated for intravenous access and the right common femoral artery was cannulated for arterial blood sampling and continuous intra-arterial blood pressure monitoring (Millar Instruments, Houston, TX, USA). A median sternotomy was performed. A catheter-tipped manometer (Millar Instruments, Houston, TX, USA) was introduced through the apex of the heart to record LV pressure. Segmental shortening in the area-at-risk (AAR) was assessed utilizing a sonometric digital ultrasonic crystal measurement system (Sonometrics Corp., London, ON, Canada) using four 2-mm digital ultrasonic probes implanted in the subepicardial layer approximately 10 mm apart within the ischemic LV area. Cardiosoft software (Sonometrics Corp., London, ON, Canada) was used for data recording (LV dP/dt, segmental shortening, arterial blood pressure, heart rate) and subsequent data analysis to determine myocardial function. Baseline hemodynamic, functional measurement, arterial blood gas analysis, and hematocrit were obtained. ABG analysis was performed every 30 min throughout the protocol. All animals received 75 mg of lidocaine as prophylaxis against ventricular dysrhythmia, as well as 80 units/kg of intravenous heparin bolus prior to occlusion of the LAD. The LAD coronary artery was occluded 3 mm distal to the origin of the second diagonal branch utilizing a Rommel tourniquet. Myocardial ischemia was confirmed visually by regional cyanosis of the myocardial surface. The Rommel tourniquet was released 60 min after the onset of acute ischemia and the myocardium was reperfused for 120 min. At the end of the reperfusion period, hemodynamic and functional measurements were recorded as described above, followed by religation of the LAD and injection of monastryl blue pigment (England Corp., Louisville, KY, USA) at a 1:150 dilution in PBS into the aortic root after placement of an aortic cross-clamp distal to the coronary arterial ostia to demarcate the AAR. The heart was rapidly excised and the entire left ventricle, including the septum, was dissected free. The LV was cut into 1 cm thick slices perpendicular to the axis of the LAD. The AAR was clearly identified by lack of blue pigment staining. Tissue from the AAR of the slice 1 cm proximal to the LV apex was isolated and divided for use in the Western blotting and immunohistochemistry. Ventricular dysrhythmia (ventricular fibrillation or pulseless ventricular tachycardia) events were recorded and treated with immediate electrical cardioversion (100–150 J, internal paddles).
Measurement of global and regional function
Global myocardial function was assessed by calculating the maximum positive first derivative of LV pressure over time (+dP/dt). Regional myocardial function was determined by using subepicardial 2 mm ultrasonic probes to calculate the percentage segment shortening (%SS), which was normalized to the baseline. Measurements were taken during a period of at least three cardiac cycles in normal sinus rhythm and then averaged. Digital data were inspected for the correct identification of end-diastole and end-systole. End-diastolic segment length (EDL) was measured at the onset of the positive dP/dt, and the end-systolic segment length (ESL) at the peak negative dP/dt. Measurements were taken at baseline (Pre) and then every 30 min (30 min after occlusion: O1, 60 min after occlusion: O2, 30 min after reperfusion: R1, 60 min after reperfusion: R2, 90 min after reperfusion: R3, 120 min after reperfusion: R4) throughout the protocol using a Sonometrics system as previously described [37, 50].
Quantification of myocardial infarct size
LV slices were immediately immersed in 1% triphenyl tetrazolium chloride (TTC, Sigma Chemical Co., St.Louis, MO, USA) in phosphate buffer (pH 7.4) at 38°C for 30 min. The infarct area (characterized by absence of staining), the non-infarcted area-at-risk (characterized by red tissue staining), and the non-ischemic portion of the LV (characterized by purple tissue staining) were sharply dissected from one another. The percentage area-at-risk was defined as: (infarct mass + non-infarct area-at-risk mass)/total LV mass × 100. Infarct size was calculated as a percentage of area-at-risk to normalize for any variation in AAR size using the following equation: (infarct mass/total mass AAR) × 100.
Western blotting
Whole-cell lysate were made from homogenized myocardial samples with a radioimmunoprecipitation assay (RIPA) buffer (Boston Bioproduct, Worcester, MA, USA) and centrifuged at 12,000×g for 10 min at 4°C to separate soluble from insoluble fractions. In the myocardial tissue lysate and blood serum, the protein concentration was measured spectrophotometrically at 595 nm wavelength with a DC protein assay kit (BioRAD, Hercules, CA, USA). Forty to eighty micrograms of total protein were fractionated by 4–20% gradient, SDS polyacrylamid gel electrophoresis (Invitrogen, San Diego, CA, USA) and transferred to PVDF membranes (Millipore, Bedford, MA, USA). Each membrane was incubated with specific antibodies as follows: anti-angiotensinogen (anti-AGT) (1:1,000 dilution R&D Systems, Minneapolis, MN, USA), anti-renin (1:1,000 dilution, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-chymase (1:1,000 dilution, abcam, Cambridge, UK), anti-angiotesin converting enzyme (anti-ACE) (1:50 dilution, abcam), anti-angiotensin II type 1 receptor (anti-AT1R) (1:500 dilution, Santa Cruz), anti-angiotensin II type 2 receptor (anti-AT2R) (1:500 dilution, Santa Cruz). The membranes were subsequently incubated for 45 min in diluted appropriate secondary antibody (Cell signaling Technology, Beverly, MA, USA). Immune complexes were visualized with the enhanced chemiluminescence detection system (Amersham, Piscataway, NJ, USA). Bands were quantified by densitometory of radioautograph films. Ponceau staining was performed to confirm equivalent protein loading.
Immunohistochemical staining
Myocardial tissue from the ischemic territory and non-ischemic territory was placed in 10% buffered formalin for 24 h, followed by paraffin mounting and sectioning into 4 μm slices. For the immunohistochemical detection of renin, ACE, Ang II, AT1R and AT2R, anti-renin (1:100 dilution, Santa Cruz), anti-angiotesin converting enzyme (anti-ACE) (1:50 dilution, abcam), anti-angiotensin II (anti-Ang II) (1:100 dilution, Peninsula Laboratories, Belmont, CA, USA), anti-angiotensin II type 1 receptor (anti-AT1R) (1:100 dilution, Santa Cruz), anti-angiotensin II type 2 receptor (anti-AT2R) (1:100 dilution, Santa Cruz) were used (4°C, overnight). Secondary labeling was achieved by using anti-mouse, rabbit, goat AlexaFluor antibodies (1:1,000 dilution, Invitrogen, OR) (30 min room temperature). Sections were mounted in Vectashield plus DAPI (Vector Laboratories, Burlingame, CA, USA). Three photographs of both AAR and NLV tissue sections of each animal (n = 6) were taken. Control studies with secondary antibodies alone were performed for convincing the specific intensities of each antibodies. The intensity of specific staining of individual sections was determined by a blinded experimenter.
Measurement of angiotensin II concentration
Ang II concentration was measured by enzyme-linked immunosorbent assay (ELISA). Serum samples of each animal in both baseline and the end of reperfusion were analyzed in a microtiter plate with enzyme immunoassay kit for Ang II according to the manufacture’s protocol (Peninsula Laboratory). Myocardial samples from each animal were homogenized in RIPA buffer (13.0 μg/μl total protein) and subjected to a tissue Ang II assay according to the manufacturer’s protocol (ALPCO Diagnostics, NH, USA).
Statistical methods
All results were expressed as mean ± standard error of the mean (SEM) and a P value of less than 0.05 was considered statistically significant (Systat, San Jose, CA, USA). Comparison between the two groups was performed using the unpaired Student’s t test and χ2 test for categorical variables. Comparison between three groups and functional data at each point was analyzed using one-way ANOVA. To identify the group or groups that differed from the others a post-hoc Dunn’s method. Immunoblots were expressed as a ratio of protein to loading band density and were analyzed after digitization and quantification of X-ray films with ImageJ 1.33 (National Institutes of Health, USA). Mean specific intensities in immunohistochemistry are calculated with each animal and analyzed with Volocity 5.2 (Improvision Inc., MA, USA).
Results
Arterial blood gas, hematocrit, core temperature, and blood sugar
No significant differences were observed between animals in arterial pH, PCO2, PO2, Hct, core temperature and blood sugar between at baseline and the end of reperfusion.
Global and regional left ventricular function
Figure 1a–d shows mean arterial blood pressure (MAP), heart rate (HR) and global systolic LV function as determined from LV dP/dt and developed LV pressure from the baseline (Pre) to the end of reperfusion (R4). No significant differences were seen between baseline and the end of reperfusion in MAP (P = 0.66), HR (P = 0.58), LV dP/dt (P = 0.56) and developed LVP (P = 0.41). Regional myocardial function in the AAR is also shown in Fig. 1e, f. The same trend was seen between baseline and the end of reperfusion in both longitudinal (P = 0.19) and horizontal (P = 0.05) axes. Table 1 shows the baseline functional data of the sham operated group.

Global and regional myocardial function. a Mean arterial blood pressure (MAP). b Heart rate (HR). c Left ventricular (LV) systolic function as determined by +LV dP/dt. d Developed LV pressure (LVP). The percentage of the segmental shortening (%SS) in longitudinal axes e and in horizontal axes f as an indicator of regional myocardial function in the area-at-risk. (*P < 0.05; one-way ANOVA). Pre pre-occlusion, O1 30 min after occlusion, O2 60 min after occlusion, R1 30 min after reperfusion, R2 60 min after reperfusion, R3 90 min after reperfusion, R4 120 min after reperfusion
Incidence of VF/VT
The incidence of ventricular fibrillation or pulseless ventricular tachycardia was measured during the experiments (1.00 ± 1.1 episodes/animal). All dysrhythmias were successfully terminated with electrical cardioversion.
Myocardial infarct size
The size of the ischemic AAR, expressed as a percentage of total LV mass was 32.3 ± 6.3% and the size of the infarct area as a percentage of total LV mass was 15.0 ± 4.9% (Fig. 2b). The size of the infracted area as a percentage of the ischemic AAR was 46.5 ± 9.7% (Fig. 2c).

Myocardial ischemic area-at-risk (AAR) and infarct size. a Left ventricular (LV) tissues after triphenyl tetrazolium chloride staining. The picture represents the first, third and fourth 1 cm thick slices cut perpendicular to the axis of the left anterior descending artery after triphenyl tetrazolium chloride staining. Three zones can be differentiated: non-ischemic area (dark red), area-at-risk (AAR; bright red) and necrotic area (pale). b Comparison between the total LV mass, AAR (32.3 ± 6.3%) and necrotic area (15.0 ± 4.9%) as a percentage of total LV mass. c Comparison between AAR and Necrotic area (46.5 ± 9.7%) as a percentage of AAR
Tissue RAS markers
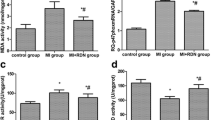
Significantly higher expression of chymase (P < 0.05), ACE (P < 0.05), AT1R (P < 0.05) and AT2R (P < 0.05) and Ang II (P < 0.05) were found in the ischemic area compared to non-ischemic area and control, whereas the expressions of AGT (P = 0.58) and renin (P = 0.38) in the myocardial tissue did not show statistical differences among groups (Fig. 3).

RAS components in myocardial tissue. a, b Western blotting of myocardial tissue for angiotensinogen (AGT), renin, chymase, angiotensin converting enzyme (ACE), angiotensin II (Ang II) type 1 receptor (AT1R) and angiotensin II type 2 receptor (AT2R), in ischemic area-at-risk (AAR; n = 6), non-ischemic left ventricle (NLV; n = 6) and sham operated group (c; n = 4). Chymase (P < 0.05), ACE (P < 0.05), AT1R (P < 0.05) and AT2R (P < 0.05) were significantly higher in AAR compared to LV and C. c Ang II level in the myocardial tissue lysate (protein concentration: 13.0 μg/μl), in AAR (n = 6), NLV (n = 6) and control (C; n = 4). (*P < 0.05; one-way ANOVA, post-hoc Dunn’s method)
Serum RAS markers
No significant differences were observed between serum levels of AGT (P = 0.57) and chymase (P = 0.71) at baseline and the end of reperfusion, whereas a significantly higher expression of serum ACE (P < 0.01) was observed at the end of reperfusion than at baseline (Fig. 4a, b). No significant difference was observed in serum Ang II concentration at baseline and at the end of reperfusion (P = 0.19) (Fig. 4c).

RAS components in serum. a, b Western blotting of serum for angiotensinogen (AGT), chymase, angiotensin converting enzyme (ACE) in the baseline (n = 6) versus the end of reperfusion (n = 6). ACE was significantly increased after reperfusion (P < 0.01). c Serum angiotensin II (Ang II) concentration in the baseline (n = 6) and the end of reperfusion (n = 6) (P = 0.19). Pre baseline, Post end of reperfusion. (*P < 0.01; unpaired Student’s t test)
Immunohistochemical staining
Specific intensities for renin, ACE, Ang II, AT1R and AT2R were detected with their respective antibodies. Significantly higher expressions of ACE (P = 0.02), Ang II (P < 0.01), AT1R (P = 0.01) and AT2R (P = 0.02) were seen in the ischemic area than non-ischemic area, whereas no significant difference in renin was seen between the ischemic and non-ischemic areas (P = 0.39) (Fig. 5).

Immunohistochemistry in myocardial tissue. a, b Immunohistochemistry for renin, angiotensin converting enzyme (ACE), angiotensin II (Ang II), Ang II type 1 receptor (AT1R) and Ang II type 2 receptor (AT2R) with the paraffin sections of non-ischemic myocardial tissue (NLV; n = 6) and ischemic area-at-risk (AAR; n = 6). The pictures represent specific intensity (bright red) for renin, ACE, Ang II, AT1R and AT2R in NLV (a, c, e, g, i) and in AAR (b, d, f, h, j). The pictures (k, l) represent controls which were labeled with secondary antibody alone (n = 6). ACE (P = 0.02), Ang II (P < 0.01), AT1R (P = 0.01) and AT2R (P = 0.02) were significantly higher in AAR than LV (*P < 0.05; unpaired Student’s t test). White bar (l) = 100 μm (a–l, d′, f′, h′, j′)
Discussion
The most significant finding in this study is that AMI-R stimulates the local RAS pathway and this leads to the synthesis of Ang II in the heart independently from the circulating RAS in the acute phase. The findings are novel in that the acute change in both the local and circulating RAS was evaluated in a clinically relevant large animal model. Recently, there have been numerous reports that the RAS plays important roles in the cardiovascular system, which are independent from an antihypertensive effect [2, 7, 17, 18, 20, 22, 24, 26, 28, 32, 35, 41, 43, 48, 49, 55, 57, 60]. Several large trials (CONSENSUS [25], SOLVD [51], ValHeFt test [6], VALLIANT [39] and CHARM [38]) have demonstrated the efficacy of ACE inhibitors and AT1R blockers (ARBs) by prolonging survival in patients with chronic heart failure, which suggests that the RAS is activated in these patients. It has been well known that modification of the RAS plays an important role in treatment of the left ventricular remodeling after myocardial ischemia and infarction [15].
As several reports have demonstrated [9, 12, 27, 40], and the results of this study suggest, the local RAS and circulating RAS have independent mechanisms. In the current study, chymase, ACE, Ang II, AT1R, and AT2R were significantly increased in the ischemic area as compared to the non-ischemic area, while serum levels of these proteins provided apparently independent behavior from myocardial expressions after 3 h of AMI-R. These results suggest that AMI-R causes direct activation of the local RAS in the ischemic myocardium in the acute period, in a part independent of the circulating RAS in a part.
It has been reported that a chymase-dependent Ang II-forming pathway is present in human cardiovascular tissues [56]. In human heart tissue, it has been reported that chymase accounts for >90% of Ang II formation. However, the biochemical and physiological roles of chymase are different in various animal species [33]. For example, in human, monkey, dog and hamster chymase cleaves the Phe8-His9 bond of angiotensin I (Ang I) and increases Ang II efficiently [4, 42, 52, 53], whereas rat chymase hydrolyzes the Tyr4-Ile5 bond of Ang I, yielding inactive fragments [31]. In the swine model, however, there are no reports about the dominant enzyme for Ang II synthesis in the heart, not to mention any detailed mechanism of swine chymase activity in vivo or the amino acid sequence of swine chymase. In this study, an increase of Ang II in the ischemic myocardial tissue involved both increased chymase and ACE locally. Further examination is required to determine the dominant enzyme responsible for cardiac Ang II synthesis and to confirm the difference between chymase-dependent and ACE-dependent Ang II in the pig. It was reported that cardiac chymase is activated in infarcted regions of a hamster myocardial infarction (MI) model during early and later phases. This activation occurred earlier and lasted longer than ACE activation [21]. This suggests that early treatment with specific drugs such as ACE inhibitors, ARBs or chymase inhibitors after AMI may decrease the myocardial remodeling resulting from activation of RAS. In this study, we evaluated the acute changes of RAS and this is meaningful in that there is an opportunity to intervene by inhibition of RAS during percutaneous coronary intervention (PCI) or coronary artery bypass grafting (CABG).
Chymase is stored in the secretory granules of mast cells and can only exert its effects after degranulation [3, 21, 30, 45]. Therefore, not only is the number of mast cells affected by acute MI, but also the rate of degranulation of mast cells may affect the production rate of Ang II after MI. Previous reports have confirmed that cardiac ischemia and reperfusion or even chronic ischemia alone can induce a degranulation of mast cells and increase the number of mast cells in infarcted myocardium [13, 21, 23]. Thus, increased chymase in the ischemic myocardium may be explained by increased delivery by mast cells. It should be noted that swine mast cells were not examined in the present study.
ACE is known to be present in the endothelium of capillaries, coronary arteries, and coronary veins [12], and is expressed on myocytes and fibroblasts in the heart [29]. ACE is produced locally, as well as taken up from the circulation [29]. In this study, local ACE levels were increased in the ischemic myocardium compared to non-ischemic myocardium and the sham, specifically in the cardiac myocytes and coronary microvessels. Circulating ACE was increased after AMI-R as well. This suggests that AMI-R leads to either the local accumulation or sequestration of circulating ACE in ischemic myocardium. In the present study, increased ACE in the ischemic myocardium was not located in the fibroblasts, but in the cardiac myocytes, which suggests that AMI-R might increase cardiac ACE in the cardiac myocytes, not in the fibroblasts. Increased ACE at the site of MI lasted for 4 weeks after occlusion of the coronary artery in a study using hamsters [21]. Clinically, this suggests that early inhibition of ACE could diminish chronic augmentation of RAS caused by AMI.
Synthesis of local Ang II in the heart occurs either extracellularly or intracellularly [29, 47, 49]. Intracellular Ang II has a variety of biologic effects, such as cell growth, gene expression, and extracellular matrix production. These functions might be the primary mediator of the chronic tissue effects of Ang II, though its exact mechanisms are not well known [29]. It has been reported that Ang II may play causative roles in cardiac remodeling and hypertrophy chronically [5, 8, 11]. Our study demonstrated that cardiac Ang II was significantly increased in the ischemic myocardium compared to non-ischemic myocardium whereas serum Ang II did not show any significant change after AMI-R. This is meaningful in that acceleration of the RAS due to AMI-R might be significant locally in cardiac RAS than in the circulating RAS during the acute period.
There is a lack of consistency between the serum levels of ACE and Ang II in this study, that is, serum ACE was significantly increased whereas serum Ang II was not changed after AMI-R. It has been suggested that serum Ang II level mainly depends on plasma renin activity and also that the serum level of Ang II is not always associated with the serum level of ACE, partly indicating the existence of the ACE-independent Ang II synthesis pathway [44].
The Ang II-receptor is divided into two major subtypes; AT1R and AT2R. AT1R has a widespread tissue distribution and mediates most of the cardiovascular Ang II actions, such as vasoconstriction and the development of some forms of cardiac hypertrophy [10, 40, 61]. The expression pattern of AT2R is more restricted. It is up-regulated in a variety of models of myocardial growth. It has been hypothesized that activation of AT2R may be antagonistic to AT1R activation. That is, AT2R exerts anti-hypertrophic effects, though detailed mechanisms of AT2R are not well known [43]. A recent article shows that chronic heart failure with cardiac hypertrophy induced by coronary ligation in a rat model causes activation of a central AT1R mechanism through augmentation of cardiac sympathetic afferent reflex [61]. Interestingly, the present study demonstrates that both AT1R and AT2R expressions are increased locally in the ischemic area compared to the non-ischemic area, suggesting activation of the local pathway early in the ischemic period. It might be that AMI-R resulted in increased AT2R in the ischemic area to compensate for the effects of increased Ang II and AT1R stimulation. Further work will be required to confirm the exact relationship between AT1R and AT2R increased by AMI-R.
RAS components have previously been shown to be expressed in cardiac myocytes [1, 47, 49, 54]. In the current study, we found that increases in Ang II, AT1R and AT2R in the ischemic myocardium were specific to cardiac myocytes alone (Fig. 5f′, h′, j′). However, other studies have shown that inflammatory cells such as macrophages also express RAS components including Ang II [36]. Further investigation will be required to verify the cell types in which RAS components are manifested.
Although this is the first report of the effect of AMI-R on cardiac RAS in a clinically relevant large animal model, several potential limitations must be taken into account. The ischemic myocardial tissue we assessed in this study might include some amount of infarcted tissue. Thus, the results in this study may reflect effects of both myocardial infarction and ischemia–reperfusion. We also realize that the immunological expression of chymase and ACE does not necessarily equate to enzymatic activity in vivo. Unfortunately, we were not able to measure the activity of these enzymes. The present study also does not investigate blood levels of renin, AT1R and AT2R, or the immunohistochemical distribution of chymase and AGT. In addition, the study also lacks the immunohistochemical studies on the sham group, but we believe that the immunoblotting results sufficiently characterize the myocardial RAS components.
In conclusion, AMI-R causes a significantly higher expression of RAS components in the acute phase in ischemic myocardium versus non-ischemic myocardium. Future work will be directed toward investigating the local RAS pathway and myocardial mechanisms after AMI-R with hope that this might yield advances in the treatment of patients after AMI.
References
Amedeo Modesti P, Zecchi-Orlandini S, Vanni S, Polidori G, Bertolozzi I, Perna AM, Formigli L, Cecioni I, Coppo M, Boddi M, Serneri GG (2002) Release of preformed Ang II from myocytes mediates angiotensinogen and ET-1 gene overexpression in vivo via AT1 receptor. J Mol Cell Cardiol 34:1491–1500
Bafford R, Sui XX, Wang G, Conte M (2006) Angiotensin II and tumor necrosis factor-alpha upregulate survivin and Kruppel-like factor 5 in smooth muscle cells: potential relevance to vein graft hyperplasia. Surgery 140:289–296
Beil WJ, Schulz M, McEuen AR, Buckley MG, Walls AF (1997) Number, fixation properties, dye-binding and protease expression of duodenal mast cells: comparisons between healthy subjects and patients with gastritis or Crohn’s disease. Histochem J 29:759–773
Caughey GH, Raymond WW, Wolters PJ (2000) Angiotensin II generation by mast cell alpha-and beta-chymases. Biochim Biophys Acta 1480
Cerbai E, Crucitti A, Sartiani L, De Paoli P, Pino R, Rodriguez ML, Gensini G, Mugelli A (2000) Long-term treatment of spontaneously hypertensive rats with losartan and electrophysiological remodeling of cardiac myocytes. Cardiovasc Res 45:388–396
Cohn JN, Tognoni G (2001) Valsartan heart failure trial investigators. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med 345:1667–1675
Cristovam PC, Arnoni CP, de Andrade MC, Casarini DE, Pereira LG, Schor N, Boim MA (2008) ACE-dependent and chymase-dependent angiotensin II generation in normal and glucose-stimulated human mesangial cells. Exp Biol Med 233:1035–1043
Dahlhof B (2001) Left ventricular hypertrophy and angiotensin II antagonists. Am J Hypert 14:174–182
Dell’Italia LJ, Husain A (2002) Dissecting the role of chymase in angiotensin II formation and heart and blood vessel diseases. Curr Opin Cardiol 17:374–379
Diniz GP, Carneiro-Ramos MS, Barreto-Chaves MLM (2009) Angiotensin type 1 receptor mediates thyroid hormone-induced cardiomyocyte hypertrophy through the Akt/GSK-3β/mTOR signaling pathway. Basic Res Cardiol 104:653–667
du Toit EF, Nabben M, Lochner A (2005) A potential role for angiotensin II in obesity induced cardiac hypertrophy and ischemic/reperfusion injury. Basic Res Cardiol 100:346–354
Endo-Mochizuki Y, Mochizuki N, Sawa H, Takada A, Okamoto H, Kawaguchi H, Nagashima K, Kitabatake A (1995) Expression of renin and angiotensin-converting enzyme in human hearts. Heart Vessels 10:285–293
Engels W, Reiters PH, Daemen MJ, Smits JF, van der Vusse GJ (1995) Transmural changes in mast cell density in rat heart after infarct induction in vivo. J Pathol 177:423–429
Ertl G, Alexander RW, Kloner RA (1983) Interactions between coronary occlusion and renin–angiotensin system in the dog. Basic Res Cardiol 78:518–533
Ertl G, Kloner RA, Alexander RW, Braunwald E (1982) Limitation of experimental infarct size by angiotensin-converting enzyme inhibitor. Circulation 65:40–48
Ertl G, Meesmann M, Kochsiek K (1985) On the mechanism of renin release during experimental myocardial ischemia. Eur J Clin Invest 15:375–381
Fulton GJ, Davies MG, Barber L, Svendsen E, Hagen PO (1998) Localized versus systemic angiotensin II receptor inhibition of intimal hyperplasia in experimental vein grafts by the specific angiotensin II receptor inhibitor L158, 809. Surgery 123:218–227
Ichihara S, Senbonmatsu T, Price E, Ichiki T, Graffney A, Inagami T (2002) Targeted deletion of angiotensin II type 2 receptor caused cardiac rupture after acute myocardial infarction. Circulation 106:2244–2249
Jalowy A, Schulz R, Dörge H, Behrends M, Heusch G (1998) Infarct size reduction by AT1-receptor blockade through a signal cascade of AT2-receptor activation, bradykinin and prostaglandins in pigs. J Am Coll Cardiol 32:1787–1796
Jin D, Takai S, Yamada M, Sakaguchi M, Kamoshita K, Ishida K, Sukenaga Y, Miyazaki M (2003) Impact of chymase inhibitor on cardiac function and survival after myocardial infarction. Cardiovasc Res 60:413–420
Jin D, Takai S, Yamada M, Sakaguchi M, Yao Y, Miyazaki M (2001) Possible roles of cardiac chymase after myocardial infarction in hamster hearts. Jpn J Pharmacol 86:203–214
Kagiyama S, Eguchi S, Frank GD, Inagami T, Zhang YC, Phillips I (2002) Angiotensin II-induced cardiac hypertrophy and hypertension are attenuated by epidermal growth factor receptor antisense. Circulation 106:909–912
Keller AW, Clancy RM, Barr ML, Marboe CC, Cannon PJ (1988) Acute reoxygenation injury in the isolated rat heart: role of resident cardiac mast cells. Circ Res 63:1044–1052
Kirimura K, Takai S, Jin D, Muramatsu M, Kishi K, Yoshikawa K, Nakabayashi M, Mino Y, Miyazaki M (2005) Role of chymase-dependent angiotensin II formation in regulating blood pressure in spontaneously hypertensive rats. Hypertens Res 28:457–464
Kjukshus J, Swedberg K, Snapinn S (1992) Effect of enalapril on long-term mortality in severe congestive heart failure. CONSENSUS Trial Group. Am J Cardiol 69:103–110
Koka V, Wang W, Huang XR, Kim-Mitsuyama S, Truong LD, Lan HY (2006) Advanced glycation end products activate a chymase-dependent angiotensin II-generating pathway in diabetic complications. Circulation 113:1353–1360
Kumar R, Baker KM, Pan J (2007) Cardiac and vascular renin–angiotensin systems. In: RM C (ed) Hypertension and hormone mechanisms. Jumana Press, Totowa, NJ, pp 23–42
Kumar R, Boim MA (2009) Diversity of pathways for intracellular angiotensin II synthesis. Curr Opin Nephro Hypertens 18:283–285
Kumar R, Singh VP, Baker KM (2009) The intracellular renin–angiotensin system in the heart. Curr Hypertens Rep 11:104–110
Lagunoff D, Pritzl P (1976) Characterization of rat mast cell granule proteins. Arch Biochem Biophys 173:554–563
Le Trong H, Neurath H, Woodbury RG (1987) Substrate specificity of the chymotrypsin-like protease in secretory granules isolated from rat mast cells. Proc Natl Acas Sci USA 84:364–367
Matsumoto C, Hayashi T, Kitada K, Yamashita C, Miyamura M, Mori T, Ukimura A, Ohkita M, Jin D, Takai S, Miyazaki M, Okada Y, Kitaura Y, Matsumura Y (2009) Chymase plays an important role in left ventricular remodeling induced by intermittent hypoxia in mice. Hypertension 54:164–171
Miyazaki M, Takai S (2000) Role of chymase on vascular proliferation. J Renin Angiotensin Aldosterone Syst 1:23–26
Nabah YN, Mateo T, Estellés R, Mata M, Zagorski J, Sarau H, Cortijo J, Morcillo EJ, Jose PJ, Sanz MJ (2004) Angiotensin II induces neutrophil accumulation in vivo through generation and release of CXC chemokines. Circulation 110:3581–3586
Newton CR, Curran B, Victorino GP (2004) Angiotensin II type 1 receptor activation increases microvascular hydraulic permeability. Surgery 136:1054–1060
Okamura A, Rakugi H, Ohishi M, Yanagitani Y, Takiuchi S, Moriguchi K, Fennessy PA, Higaki J, Ogihara T (1999) Upregulation of renin–angiotensin system during differentiation of monocytes to macrophages. J Hypertens 17:537–545
Osipov RM, Bianchi C, Clements RT, Feng J, Liu Y, Xu SH, Robich MP, Wagstaff J, Sellke FW (2009) Thrombin fragment (TP508) decreases myocardial infarction and apoptosis after ischemia reperfusion injury. Ann Thorac Surg 87:786–793
Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJ, Michelson EL, Olofsson B, Ostergren J, Yusuf S, Pocock S (2003) CHARM Investigators and Committees. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet 362:759–766
Pfeffer MA, McMurray JJ, Velazquez EJ, Rouleau JL, Køber L, Maggioni AP, Solomon SD, Swedberg K, Van de Werf F, White H, Leimberger JD, Henis M, Edwards S, Zelenkofske S, Sellers MA, Califf RM (2003) Valsartan in Acute Myocardial Infarction Trial Investigators. Valsartan, captopril, or both in myocardial infarction complicated by heart failure, left ventricular dysfunction, or both. N Engl J Med 349:1893–1906
Preckel B, Schlack W, Gonzalez M, Obal D, Barthel H, Thamer V (2000) Influence of the angiotensin II AT1 receptor antagonist irbesartan on ischemia/reperfusion injury in the dog hear. Basic Res Cardiol 95:404–412
Prossed HCG, Forster ME, Richards AM, Pemberton CJ (2009) Cardiac chymase converts rat proAngiotensin-12 (PA12) to angiotensin II: effects of PA12 upon cardiac haemodynamics. Cardiovasc Res 82:40–50
Reilly CF, Tewksbury DA, Schechter NM, Travis J (1982) Rapid conversion of angiotensin I to angiotensin II by neutrophil and mast cell proteinases. J Biol Chem 257:8619–8622
Ritter O, Schuh K, Brede M, Röthlein N, Burkard N, Hein L, Neyses L (2003) AT2-receptor activation regulates myocardial eNOS expression via the calcineurin-NF-AT pathway. FASEB J 17:283–285
Rosendorff C (1996) The renin–angiotensin system and vascular hypertrophy. J Am Coll Cardiol 28:803–812
Sayama S, Iozzo RV, Lazarus GS, Schechter NM (1987) Human skin chymotrypsin-like proteinase chym. J Biol Chem 262:6808–6815
Saygili E, Rana OR, Meyer C, Gemein C, Andrzejewski MG, Ludwig A, Weber C, Schotten U, Krüttgen A, Weis J, Schwinger RH, Mischke K, Rassaf T, Kelm M, Schauerte P (2009) The angiotensin-calcineurin-NFAT pathway mediates stretch-induced up-regulation of matrix metalloproteinases-2/-9 in atrial myocytes. Basic Res Cardiol 104:48–435
Singh VP, Le B, Bhat VB, Baker KM, Kumar R (2007) High-glucose-induced regulation of intracellular ANG II synthesis and nuclear redistribution in cardiac myocytes. Am J Physiol Heart Circ Physiol 293:939–948
Singh VP, Baker KM, Kumar R (2008) Activation of the intracellular renin–angiotensin system in cardiac fibroblasts by high glucose: role in extracellular matrix production. Am J Physiol Heart Circ Physiol 294:1675–1684
Singh VP, Le B, Khode R, Baker KM, Kumar R (2008) Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes 57:3297–3306
Sodha NR, Clements RT, Feng J, Liu Y, Bianchi C, Horvath EM, Szabo C, Sellke FW (2008) The effects of therapeutic sulfide on myocardial apoptosis in response to ischemia-reperfusion injury. Eur J Cardiothorac Surg 33:906–913
SOLVD (1992) Effect of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fractions. The SOLVD Investigattors. N Engl J Med 327:685–691
Takai S, Shiota N, Kobayashi S, Matsumura E, Miyazaki M (1997) Induction of chymase that forms angiotensin II in the monkey atherosclerotic aorta. FEBS Lett 412:86–90
Takai S, Shiota N, Yamamoto D, Okunishi H, Miyazaki M (1996) Purification and characterization of angiotensin II-generating chymase from hamster cheek pouch. Life Sci 58:591–597
Tsai CT, Lai LP, Hwang JJ, Chen WP, Chiang FT, Hsu KL, Tseng CD, Tseng YZ, Lin JL (2008) Renin–angiotensin system component expression in the HL-1 atrial cell line and in a pig model of atrial fibrillation. J Hypertens 26:570–582
Urata H, Boehm KD, Philip A, Kinoshita A, Gabrovsek J, Bumpus FM, Husain A (1993) Cellular localization and regional distribution of an angiotensin II-forming chymase in the heart. J Clin Invest 91:1269–1281
Urata H, Kinoshita A, Misono KS, Bumpus FM, Husain A (1990) Indentification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J Biol Chem 265:22348–22357
Utsunomiya H, Nakamura M, Kakubo K, Inagami T, Tamura M (2005) Angiotensin II AT2 receptor localization in cardiovascular tissues by its antibody developed in AT2 gene-deleted mice. Regul Pept 126:155–161
Weidenbach R, Schulz R, Gres P, Behrends M, Post H, Heusch G (2000) Enhanced reduction of myocardial infarct size by combined ACE inhibition and AT1 receptor antagonism. Br J Pharmacol 131:138–144
Wenzel S, Tastan I, Abdallah Y, Schreckenberg R, Schlüter KD (2009) Aldosterone improves contractile function of adult rat ventricular cardiomyocytes in a non-acute way: potential relationship to the calcium paradox of aldosteronism. Basic Res Cardiol 105:247–256
Yam FK, Akers WS, Ferraris VA, Smith K, Ramaiah C, Camp P, Flynn JD (2006) Interventions to improve guideline compliance following coronary artery bypass grafting. Surgery 140:541–552
Zhu GQ, Zucker IH, Wang W (2002) Central AT1 receptor involved in the enhanced cardiac sympathetic afferent reflex in rats with chronic heart failure. Basic Res Cardiol 97:320–326
Acknowledgments
We thank the staff of the Animal Research Facility and Jessinia Urrea at the Beth Israel Deaconess Medical Center for their efforts.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Oyamada, S., Bianchi, C., Takai, S. et al. Impact of acute myocardial ischemia reperfusion on the tissue and blood-borne renin–angiotensin system. Basic Res Cardiol 105, 513–522 (2010). https://doi.org/10.1007/s00395-010-0093-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00395-010-0093-4