
Abstract
Purpose
Bone morphogenetic proteins (BMP) have been shown to play crucial roles in not only lung and heart development, but also in the pathogenesis of pulmonary vascular remodeling in pulmonary hypertension (PH). We therefore hypothesized that BMP signaling could be altered in nitrofen-induced congenital diaphragmatic hernia (CDH) and associated PH.
Methods
Pregnant rats were exposed to either 100 mg nitrofen or vehicle on embryonic day (E) 9.5. On E17 and E21, fetuses were delivered by cesarean section, killed and checked for left-sided CDH. The tissue was then harvested for pathobiological evaluation.
Results
In nitrofen-induced CDH, pulmonary expressions of BMP4, BMP receptor (BMPR) type 2 and Id1 decreased on E17 and E21. On E17, pulmonary gremlin-1 expression increased, while BMP7 decreased. In the lungs, Id1 expression was correlated to BMP4 and BMPR2 and inversely correlated to gremlin-1 expression. Myocardial expressions of BMPR2, BMPR1A, BMP7 and SERCA-2A decreased, while gremlin-1 and noggin expressions increased on E17. On E21, myocardial expressions of Id1 and SERCA-2A decreased, while gremlin-1 expression increased. Moreover, BMPR2 and BMPR1A expressions were correlated to SERCA-2A expression and inversely correlated to pro-apoptotic Bax/Bcl2 ratio within the myocardium.
Conclusion
Downregulation of BMP signaling seems to contribute to pulmonary and myocardial anomalies observed in nitrofen-induced CDH.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Congenital diaphragmatic hernia (CDH) is a life-threatening cause of severe respiratory failure in the newborn, affecting 1/4000 live births. CDH is caused by a defect in the integrity of the diaphragm that allows the abdominal content to ascend into the thorax, thereby compromising in utero lung development. Despite recent advances in prenatal care and resuscitation, isolated CDH has a persistently high mortality rate (30–40 %) [1, 2], mainly due to co-existing abnormalities such as pulmonary hypoplasia associated with abnormal pulmonary vascular development and persistent pulmonary hypertension of the newborn (PPHN), intestinal malrotation and left heart hypoplasia [3]. Cardiovascular malformations have been reported in 11–15 % of the newborns with isolated CDH and associated with poorer prognosis than either malformation alone [4]. Despite recent findings in experimental and human CDH, the pathogenesis of pulmonary hypoplasia, pulmonary vascular remodeling and cardiovascular malformations associated with CDH remains poorly understood.
During development and organogenesis, key growth factors such as bone morphogenetic proteins (BMPs) have been shown to drive organ and tissue development. The importance of BMP signaling has been highlighted by the fact that deletion in BMP signaling (through BMP receptor (BMPR) type 2 (BMPR2) homozygous mutant) results in early embryonic lethality in mice due to the failure in gastrulation [5]. Later during development, BMPs have also been shown to be involved in organogenesis, particularly that of the lung, heart and kidney [6, 7].
In the lungs, BMPs have been shown to play critical roles in airway branching morphogenesis in the early development stages [8, 9] and also later in pulmonary septal and vascular development [10]. Overexpression of a BMP antagonist, the gremlin, in the distal airway epithelium has been shown to cause disruption of normal airway patterning [11, 12]. Moreover, mutations in the gene encoding BMPR2 associated with decreased BMPR2 expression have been reported in the heritable form of pulmonary arterial hypertension (PAH) as implicated in the pathogenesis of pulmonary vascular remodeling [13]. In the developing heart, defective BMP signaling has also been shown to disrupt the intricate steps of cardiac formation (resulting in congenital heart defects [14, 15]), probably acting on myocardial differentiation [16]. We, therefore, hypothesized that the BMP/BMPR2 signaling pathway, which seems to play crucial roles in normal lung and heart development, may be altered in CDH and possibly implicated in fatal anomalies associated with CDH and PPHN.
In the present study, we evaluated the pulmonary gene expressions of the main members of BMP signaling, including BMP receptors (BMPR1A and BMPR2), BMP agonists (BMP4 and BMP7) and BMP antagonists (gremlin and noggin), as well as the expression and/or the activation of several targets of BMP signaling (e.g., the gene expression of the inhibitor of DNA binding 1 (Id1) and activation of Smad1/5/8 signaling pathway) on embryonic day (E) 17 and/or 21 of lung development in a nitrofen-induced experimental rat model of CDH. We also explored this signaling pathway in the hypoplastic hearts in this experimental model of CDH.
Methods
All procedures and protocols were approved by the institutional committee on animal welfare at the Université Libre de Bruxelles (Brussels, Belgium).
Animal model
Pregnant Sprague–Dawley rats (Janvier, Saint-Barthevin, France) were gavage fed with 100 mg herbicide nitrofen (2,4-dichloro-4′-nitrodiphenyl ether; Fluka, Deisenhofen, Germany) dissolved in 1.5 mL olive oil vehicle at E9.5 (term, 22 days). Nitrofen is not toxic to adult rats, but induces CDH in the offspring. The incidence of CDH varies depending on the timing of nitrofen administration [17]. Control rats received olive oil vehicle only. Pregnant Sprague–Dawley rats were randomized into two groups: control and nitrofen administered. On E17 and E21, the animals were anesthetized and fetuses were harvested by cesarean section and assessed for left-sided CDH development in the nitrofen litters. Because not all fetuses develop CDH, only those that developed CDH in the nitrofen litters were selected and further analyzed. Fetuses were then divided into two groups: control and nitrofen-induced CDH. Fetuses were weighed and killed by exsanguination. Pulmonary and myocardial tissues were immediately harvested and weighed. Lungs (n = 15 in nitrofen-induced CDH and n = 17 in control group from two different litters, on both E17 and E21) and heart (n = 15 in nitrofen-induced CDH and n = 17 in control group from two different litters, on both E17 and E21) were snap-frozen in liquid nitrogen and stored at −80 °C for pathobiological evaluation. After overnight fixation, lungs (n = 4 in each group on E21) were embedded in toto in paraffin for histopathological evaluation.
Lung morphometry
Serial sections, 5 μm in thickness, were taken along the longitudinal axis of the lung lobes and stained with hematoxylin and eosin (H&E). Morphometrical analysis was performed estimating the mean wall transection length (Lmw) [18, 19] and the radial alveolar count (RAC) [20, 21]. All counts were performed by two independent investigators in a blinded manner. The mean value was used for analysis.
All measurements were performed with a Zeiss AXIOPLAN light microscope (Carl Zeiss, Germany) at a total magnification of 100×. The total surface of each lung section was virtually divided in up to 20 random non-overlapping fields for morphometric analysis. Parameters were measured in the lung parenchyma focusing on the respiratory airways as previously described by Roubliova et al. [18]. The radial alveolar count (RAC) method was applied according to the method previously used by Emery and Mithal [20].
To assess pulmonary arterial remodeling, pulmonary arterial morphometry was performed as previously described [22]. Only small pulmonary arteries with an external diameter (ED) <100 μm and a complete muscular coat were measured. Medial thickness (MT) was related to arterial size by the following formula: %MT = (MT/ED) × 100 and was measured by counting at least 50 pulmonary arteries per lung lobe from each fetus. Pulmonary vascular density (number of pulmonary arteries with ED < 100 μm/mm²) was evaluated using at least 50 microscopic fields (in 100× total magnification) of pulmonary sections randomly selected.
Lung BMPR2 and gremlin-1 immunolocalization
Lung immunostaining was performed as previously described [23]. Briefly, 5 μm lung sections were dewaxed and progressively rehydrated. For antigen retrieval, sections were incubated in target retrieval buffer (Dako, Glostrup, Denmark) and heated in a bath for 20 min at 95 °C. Endogenous peroxidase activity was quenched with hydrogen peroxide in PBS (1 %, vol/vol) for 10 min, and the sections were blocked by incubation with bovine serum albumin in PBS (3 %, vol/vol) for 30 min. Sections were allowed to react overnight at 4 °C with goat anti-human BMPR2 antibody or goat anti-mouse gremlin-1 antibody (diluted 1:100 in PBS; R&D Systems, Abingdon, UK). Sections were then incubated with biotinylated anti-rabbit IgG (Dako, Le Perray en Yvelines, France) and subsequently with streptavidin–peroxidase (Dako). Antibody binding was detected with a liquid diaminobenzidine (DAB) substrate kit (AbCvs Biology, Paris, France). The appearance of a brown reaction product was observed under a light microscope. Nuclei were counterstained with hematoxylin and mounted. Negative controls run without the primary antibody were tested.
RNA extraction and reverse transcription
Total RNA was extracted from snap-frozen pulmonary and myocardial tissue using TRIzol reagent (Invitrogen, Merelbeke, Belgium) and purified with RNeasy Mini kit (QIAGEN, Hilden, Germany), according to the manufacturer’s instructions. RNA concentration was determined by standard spectrophotometric technique. After electrophoresis, RNA integrity was assessed by visual inspection of ethidium bromide-stained 1.5 % agarose gels. Reverse transcription was performed using random hexamer primers and Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA, USA), according to the manufacturer’s instructions.
Real-time quantitative polymerase chain reaction
For real-time quantitative polymerase chain reaction (RTQ–PCR), sense and antisense primers were designed using Primer3 program for rattus norvegicus BMPR1A and BMPR2, BMP4 and BMP7, gremlin-1, noggin, Id1, Bax, Bcl-2, sarcoplasmic/endoplasmic reticulum calcium ATPase-2 (SERCA-2A) and hypoxanthine phosphoribosyltransferase (HPRT) mRNA sequences (Table 1). To avoid inappropriate amplification of residual genomic DNA, intron-spanning primers were selected. A BLAST analysis was performed to check if primers only matched at the sequence of interest.
For each sample, amplification reaction was performed in triplicate using SYBR Green PCR Master Mix (Quanta Biosciences, Gaithersburg, MD, USA), specific sense and anti-sense primers and diluted template cDNA. Result analysis was performed using an iCycler System (BioRad Laboratories, Nazareth Eke, Belgium). Relative quantification was achieved with the comparative \(2^{-\Updelta \Updelta C_{\text{t}}}\) method by normalization with the housekeeping gene (HPRT). Results were expressed as relative fold increase over the mean value of relative mRNA expression of control group arbitrarily fixed to 1.
Immunoblotting for BMP signaling pathway
Proteins were extracted from snap-frozen pulmonary tissue samples by homogenization in an appropriate amount of ice-cold homogenizing lysis buffer [Tris–HCl pH 7.4, NaCl, NaF, sodium pyrophosphate (all at 25 mM), sodium vanadate (1 mM), EDTA, EGTA (both at 2.5 mM), phenylmethylsulfonyl fluoride (1 mM), aprotinine, leupeptine (both at 5 μg mL−1), SDS, deoxycholate and NP-40 (all at 0.50 %)], as previously described [13]. The homogenates were centrifuged at 4 °C and the supernatants were collected. After determination of the protein concentration using the method of Bradford [24], protein extracts (100 μg) were resolved on 4–12 % NuPage Bis–Tris gels (Invitrogen, Carlsbad, CA, USA) and electro-transferred to nitrocellulose membranes (BioRad, Hercule, CA, USA). After blocking with 5 % non-fat milk in TBS [Tris–HCl (pH 8.0); NaCl 150 mM] for 1 h at room temperature, the membranes were incubated successively with polyclonal rabbit anti-human phospho-Smad1 (Ser463/465)/Smad5 (Ser463/465)/Smad8 (Ser426/428) (1:1000; Cell Signaling Technology Inc., Danvers, MA, USA) and polyclonal goat anti-human total Smad1/5/8 (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4 °C overnight with rocking. After incubation with secondary HRP-conjugated anti-rabbit and anti-goat antibody, respectively (1:25000; ThermoScientific, Rockford, IL, USA), immunoreactive bands were detected using SuperSignal® WestPico Chemiluminescent substrate (ThermoScientific, Rockford, IL, USA) and quantified with a computed optical method using the Bio 1D software (Vilber Lourmat, France). Relative quantification was performed by normalization with β-actin (Sigma-Aldrich, St Louis, MO, USA).
Statistical analysis
Data are reported as mean ± SEM. Statistical analyses were performed using StatView 5.0 software. Comparisons between groups were performed using one-way analysis of variance (one-way ANOVA). When the F ratio of this analysis resulted in a P value of <0.05 critical value, comparisons were made with a modified two-tailed Student’s t test. P values <0.05 were considered to be statistically significant.
Results
Experimental model—lung architecture, pulmonary vascular density and morphometry
In nitrofen-exposed litters, left-sided CDH was found in 67 % of the fetuses. The lung-to-body weight ratio (LW/BW), a crude indicator of lung hypoplasia, was lower by 28 and 12 % on E17 and E21, respectively, in nitrofen-induced CDH fetuses compared to controls (P < 0.05 at both embryonic days). The heart-to-body weight ratio (HW/BW), assessing the heart hypoplasia, was lower in nitrofen-induced CDH fetuses compared to controls on E17 and E21, by 35 and 8 %, respectively (P < 0.05 at each embryonic day). There was a correlation between lung- and heart-to-body weight ratios (R = 0.274; P = 0.028).
As previously reported, thickness of the alveolar septa (assessed by Lmw) increased, while the radio-alveolar count (RAC) decreased on E21 in nitrofen-induced CDH compared to control fetuses. Pulmonary vessels were less abundant (3 ± 0 vs. 2 ± 0 pulmonary arteries/pulmonary parenchyma section at 100× total magnitude, P < 0.001) and presented with decreased external diameter and increased pulmonary medial thickness in fetuses with nitrofen-induced CDH compared to controls.
Pulmonary hypoplasia and vascular remodeling—BMPR2 signaling and apoptosis
Nitrofen-induced CDH, characterized by lung hypoplasia and vascular remodeling, was associated on E17 with decreased pulmonary gene expressions in two BMP agonists, BMP4 and BMP7, while only pulmonary BMP4 gene expression decreased on E21 (Fig. 1a, b). Gene expression in gremlin-1, a BMP antagonist, increased on E17, while it did not change between two groups on E21 (Fig. 1c). There was no change in gene expression in noggin, another BMP antagonist, on E17 and E21 (Fig. 1d). As illustrated in Fig. 2c, f, gremlin-1 was expressed by lung epithelial cells and also by vascular cells in pulmonary vessels in both groups.

Pulmonary expressions of BMP agonists and antagonists. Relative lung mRNA content for the bone morphogenetic protein signaling pathway, including the agonists: bone morphogenetic protein 4 (BMP4) (a) and bone morphogenetic protein 7 (BMP7) (b) and the antagonists: gremlin-1 (c) and noggin (d) on embryonic days 17 (E17) and 21 (E21) in control (n = 9, white bars) and nitrofen-induced congenital diaphragmatic hernia (nitrofen-CDH, n = 7, black bars) groups. Values are expressed as mean ± SEM. *0.01 < P < 0.05 nitrofen-induced congenital diaphragmatic hernia (CDH) versus control lungs. ††0.001 < P < 0.01, ††† P < 0.001, E21 versus E17 fetuses in the same group

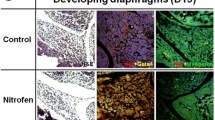
Lung and pulmonary vascular BMPR2 and gremlin-1 immunolocalization. Representative hematoxylin and eosin-stained pulmonary sections (a) and pulmonary arterioles (d) obtained from controls and fetuses with nitrofen-induced congenital diaphragmatic hernia (nitrofen-CDH) at 400-fold total magnification, showing thickened mesenchyme (a) and pulmonary arteriolar remodeling (d) in nitrofen-induced congenital diaphragmatic hernia. Representative slides of BMPR2 immunostaining in lung sections obtained from control and nitrofen-induced CDH fetuses (b, e). BMPR2 immunostaining is detected in the epithelium and in the pulmonary vessels in the lungs obtained from control and nitrofen-induced CDH fetuses. Representative slides of gremlin-1 immunostaining in lung sections obtained from control and nitrofen-induced CDH fetuses (c, f). Gremlin-1 immunostaining is detected in the epithelium and in the pulmonary vessels in the lungs obtained from control and nitrofen-induced CDH fetuses. Scale bars: 100 μm
Decreased pulmonary gene expression in BMPR2 was observed on E17 and E21 in nitrofen-induced CDH (Fig. 3a), while pulmonary BMPR1A gene expression did not change (Fig. 3b). As illustrated in Fig. 2b, e, BMPR2 staining was present in the epithelium and also in the medial layer of the pulmonary vessels. Pulmonary gene expression of the DNA binding protein inhibitor (Id) 1, a BMP-responsive gene which encodes a small helix-loop-helix inhibitor of differentiation protein usually used to read out the BMP signaling, decreased on E17 and E21 during nitrofen-induced CDH development (Fig. 3c), suggesting that the activation of the BMP signaling was decreased in the lungs of nitrofen-induced CDH fetuses. Moreover, pulmonary Id1 expression was correlated to pulmonary BMP4 and BMPR2, and inversely correlated to pulmonary gremlin-1 expression (Fig. 4a–c). Pulmonary activation (by phosphorylation) of Smad1/5/8 signaling pathway decreased on E21 in nitrofen-induced CDH (Fig. 3e), indicating decreased activation of BMP signaling.

Pulmonary expressions of BMP receptors and pro-apoptotic Bax-to-Bcl-2 ratio. Relative lung mRNA content for the bone morphogenetic protein receptor signaling pathway, including the bone morphogenetic protein receptor type 2 (BMPR2) (a) and the bone morphogenetic protein receptor type 1A (BMPR1A) (b) and for the inhibitor of DNA binding (Id1) (c) on embryonic days 17 (E17) and 21 (E21) in control (n = 9, white bars) and nitrofen-induced congenital diaphragmatic hernia (nitrofen-CDH, n = 7, black bars) groups. Pro-apoptotic ratio of mitochondrial members of Bcl-2 protein family, the Bax-to-Bcl-2 ratio (d) on embryonic days 17 (E17) and 21 (E21) in control (n = 9, white bars) and nitrofen-induced congenital diaphragmatic hernia (nitrofen-CDH, n = 7, black bars) groups. Pulmonary activation (by phosphorylation) of Smad1/5/8 signaling. Representative Western blots for phosphorylated Smad1/5/8, total Smad1/5/8 and beta-actin expressions. Beta-actin expression evaluation was performed to show equal loading. Densitometry of relative phospho-Smad1/5/8 related to total Smad1/5/8 protein expression on embryonic day 21 (E21) in total lung homogenates from control (n = 8, white bars) and nitrofen-induced congenital diaphragmatic hernia (nitrofen-CDH, n = 8, black bars) groups (e). Values are expressed as mean ± SEM. *0.01 < P < 0.05; **0.001 < P < 0.01 nitrofen-induced congenital diaphragmatic hernia (CDH) versus control lungs. ††0.001 < P < 0.01, ††† P < 0.001, E21 versus E17 fetuses in the same group

Correlations between lung Id1 mRNA expression and lung mRNA contents of bone morphogenetic protein 4 (BMP4; a), bone morphogenetic protein receptor type 2 (BMPR2; b) and gremlin-1 (c) on E17 and E21
The pro-apoptotic Bax-to-Bcl-2 ratio decreased on E21 in the lung of fetuses with nitrofen-induced CDH, while it did not change on E17 (Fig. 3d). Nitrofen administration decreased, on E21, pulmonary gene expression of pro-apoptotic Bax mitochondrial member (1.22 ± 0.12 vs. 0.32 ± 0.03; P < 0.001), while anti-apoptotic Bcl-2 gene expression did not change (1.12 ± 0.32 vs. 1.39 ± 0.41; NS). No correlation was found between the pulmonary expressions of BMP/BMPR2 signaling members and this pro-apoptotic Bax/Bcl2 ratio.
Congenital heart hypoplasia—BMPR2 signaling, apoptosis and calcium handling
As illustrated in Fig. 5, nitrofen-induced heart hypoplasia was associated with decreased myocardial gene expressions in BMPR2 and BMPR1A on E17 (Fig. 5a, b). Myocardial gene expression in BMP7, a BMPR2 agonist, decreased on E17 (1.00 ± 0.03 vs. 0.64 ± 0.07; P < 0.05), while BMP4 expression did not change (1.00 ± 0.19 vs. 0.99 ± 0.15; NS). No changes in myocardial BMPR2 (Fig. 5a), BMPR1A (Fig. 5b), BMP4 and BMP7 were seen on E21. Myocardial gene expressions of BMP antagonists (gremlin-1 and noggin) increased on E17 (Fig. 5c, d), while, on E21, only myocardial gremlin-1 gene expression increased (Fig. 5c). In the nitrofen-induced CDH group, myocardial Id1 gene expression decreased on E21, but not on E17 (Fig. 5e), suggesting decreased activation of BMPR2 signaling on E21.

Myocardial expressions of genes related to BMP signaling pathway and genes implicated in calcium-dependent myocardial contractility. Relative myocardial mRNA content for the bone morphogenetic protein receptor signaling pathway, including the bone morphogenetic protein receptor type 2 (BMPR2) (a), the bone morphogenetic protein receptor type 1A (BMPR1A) (b), the antagonists, gremlin-1 (c) and noggin (d), and the inhibitor of DNA binding (Id1) (e) on embryonic days 17 (E17) and 21 (E21) in control (n = 9, white bars) and nitrofen-induced congenital diaphragmatic hernia (nitrofen-CDH, n = 7, black bars) groups. Myocardial mRNA content for the sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA-2A) (f) on embryonic days 17 (E17) and 21 (E21) in control (n = 9, white bars) and nitrofen-induced congenital diaphragmatic hernia (nitrofen-CDH, n = 7, black bars) groups. Values are expressed as mean ± SEM. *0.01 < P < 0.05; **0.001 < P < 0.01 nitrofen-induced congenital diaphragmatic hernia (CDH) versus control hearts. ††0.001 < P < 0.01, ††† P < 0.001, E21 versus E17 fetuses in the same group
In the hypoplastic heart of fetuses with nitrofen-induced CDH, the myocardial pro-apoptotic Bax/Bcl-2 ratio did not change on E17 and E21 (1.00 ± 0.26 vs. 0.81 ± 0.25 on E17 and 1.01 ± 0.18 vs. 0.99 ± 0.17 on E21). The myocardial gene expression of SERCA-2A, an ATPase implicated in calcium-dependent contractility in cardiomyocytes, decreased on E17 and E21 (Fig. 5f). Moreover, myocardial SERCA-2A expression was correlated to BMPR2 and BMPR1A expressions (Fig. 6a, b) and myocardial pro-apoptotic Bax/Bcl2 ratio was inversely correlated to BMPR2 and BMPR1A expressions (Fig. 6c, d), suggesting possible link between the BMP/BMPR2 signaling and the calcium-dependent myocardial contraction and the activation of apoptotic processes within the myocardium.

Correlations between heart sarcoplasmic/endoplasmic reticulum calcium ATPase 2 (SERCA-2A) mRNA expression and heart mRNA contents of bone morphogenetic protein receptor type 2 (BMPR2; a) and bone morphogenetic protein receptor type 1A (BMPR1A; b). Inverse correlations between heart expression of pro-apoptotic Bax/Bcl-2 ratio and heart expressions of bone morphogenetic protein receptor type 2 (BMPR2; c) and bone morphogenetic protein receptor type 1A (BMPR1A; d) on E17 and E21
Discussion
The present results show downregulated BMP/BMPR2 signaling in the lungs and heart of fetuses with congenital diaphragmatic hernia (CDH) experimentally induced by the administration of nitrofen in rats.
BMP signaling has been shown to play crucial roles in regulating early lung branching morphogenesis [25, 26] via the reciprocal interactions between epithelial and mesenchymal cells and also later during lung maturation [10], promoting the survival of progenitor cells mediating surfactant production and facilitating fluid clearance from the air sacs of neonatal lungs [27]. Inhibiting BMP signaling in mouse lung epithelial cells in either early lung organogenesis or late gestation results in retardation of lung branching morphogenesis, and reduced epithelial cell proliferation and differentiation [9], leading to neonatal respiratory distress phenotype accompanied by collapsed lungs with altered function and structure of the airways [27]. The present results show that the BMP signaling was downregulated in the hypoplastic lungs with nitrofen-induced CDH during the canalicular and the saccular stages of embryonic lung development. These results are consistent with studies reporting reduced pulmonary expression of BMP4 [28] and Smad1 [29] in the hypoplastic lungs with nitrofen-induced CDH, suggesting that a defect in BMP signaling could be one of the mechanisms responsible for the alteration in early branching morphogenesis and lung maturation in this experimental model of CDH. In contrast, similar activation of downstream BMPR2 signaling pathway has been recently shown (the Smad1/5/8 signaling), in lungs with nitrofen-induced CDH compared to normal lungs [30]. The reasons for these apparent discrepancies are not clear. However, the same study also failed to find any alteration of the angiopoetin/Tie2 signaling, which has already been described as altered in experimental CDH [31, 32] and related to abnormalities in BMPR2 expression in patients with pulmonary arterial hypertension (PAH) [33].
Pulmonary expressions of BMP agonists, such as BMP4 and BMP7, were decreased during critical periods of lung development in nitrofen-induced CDH, together with early increased expression of BMP antagonist gremlin-1. Previous studies have shown that adding BMP4 to organ cultures of the whole embryonic lung promotes branching morphogenesis [34]. In contrast, decreased BMP4 expression [8] and overexpression in BMP antagonists within the epithelium [9, 11] have been shown to result both in smaller lungs with dilated terminal saccules and altered epithelial cell proliferation and differentiation leading to disrupted distal airway formation, which do not support gas exchange and result in neonatal respiratory failure [27]. Moreover, transgenic mice deficient in gremlin-1 presented with dramatic pulmonary abnormalities [35]. Increased pulmonary levels of gremlin-1 have been recently suggested as being implicated in pulmonary vascular remodeling in hypoxic pulmonary hypertension [36]. This suggests that the alteration of the balance between BMP agonists and antagonists which has been shown to be crucial during lung development [25] could be related to lung hypoplasia and defect in lung branching and also pulmonary vascular remodeling in experimental CDH induced by nitrofen exposure in rats.
In lungs with nitrofen-induced CDH, BMPR2 expression decreased, together with decreased activation of Smad1/5/8 signaling and decreased expression of BMP-responsive gene Id1, suggesting decreased expression and function of BMP signaling. Alterations in the pulmonary expression/function of BMPR2 could also be related to vascular abnormalities observed in nitrofen-induced CDH. Indeed, BMP/BMPR2 signaling has been shown to contribute to the maintenance of pulmonary vascular structure and function [37, 38]. Mutations in BMPR2 (associated with decreased BMPR2 expression) have been described in PAH patients [39]. Downregulated BMP signaling has been shown to be implicated in the pathogenesis of PAH, mainly in pulmonary arterial remodeling [13, 36]. This suggests that the decreased BMPR2 expression in the lungs with nitrofen-induced CDH could be related to defect in lung maturation and also in vascular remodeling. BMPR1A has been shown to be a key component in BMP-regulated lung development [40]. However, in nitrofen-induced CDH, BMPR1A expression does not seem to be affected in lungs.
Disturbed interplay between cell proliferation and apoptosis may contribute to the observed hypoplasia in the lung explants exposed to nitrofen [41], with associated increased activation of the apoptotic processes [42] or not [41, 43]. Our results did not show activation of apoptosis (assessed by pro-apoptotic Bax/Bcl-2 ratio evaluation) during the canalicular stage of embryonic lung development in nitrofen-induced CDH. However, the activation of apoptosis was decreased during late lung development in nitrofen-induced CDH. The reasons for these apparent discrepant results are not clear, even if this decreased apoptosis could participate in the thickening of the interalveolar septa and the pulmonary vascular remodeling observed in this experimental model.
Nitrofen exposure is also known to induce a variety of congenital cardiovascular malformations, mimicking congenital heart malformations observed in patients with CDH. Defects in cardiac morphogenesis have been reported in transgenic mice expressing BMPR2-mutant [44, 45] and cardiac defects; particularly, those involving the endocardial cushion have been identified in patients with congenital heart disease associated with BMPR2 mutation [46]. Endocardial BMPR2 expression is required for septal formation and valvulogenesis, and mesenchymal BMPR2 expression in the outflow tract cushion is required for proper positioning of the aorta [14]. Evaluating cardiac function in rat fetuses is difficult because of reduced dimensions of the pups, but we confirmed heart hypoplasia and decreased myocardial expression of SERCA-2A [47], a gene developmentally regulated, sensible to load conditions and evaluating myocardial maturity. Moreover, correlations found between myocardial BMPR2 and BMPR1A expressions and myocardial SERCA-2A expression and pro-apoptotic Bax/Bcl2 ratio strongly suggest the functional impact of this BMP/BMPR2 signaling pathway in the heart.
Although the mechanisms by which nitrofen induces CDH is not fully understood, a dual-hit hypothesis explaining the development of CDH and lung hypoplasia has been proposed in this experimental model [41]. The early retardation in lung development that occurs before the development of the diaphragmatic defect (detected as early as E13–14 during lung development [48, 49]) seems to be directly mediated by nitrofen [41, 50], whereas the later gestational increase in lung hypoplasia seems to be caused by mechanical compression from herniated liver into the chest cavity [49]. In the present work, we cannot exclude the toxic effect of nitrofen exposure on the expression of BMP signaling members. However, altered expression observed during late lung development suggests altered BMP signaling independently of nitrofen. For these reasons, BMP signaling should be further investigated in the lungs and heart from other experimental or human CDH.
The present results support the notion of downregulated BMP/BMPR2 signaling in lung hypoplasia and pulmonary vascular remodeling associated with nitrofen-induced experimental left-sided CDH. In the heart of fetuses with nitrofen-induced CDH, activation of BMP/BMPR2 signaling is also reduced. This suggests that BMP signaling plays crucial roles in lung and heart hypoplasia associated with nitrofen-induced CDH and also in pulmonary vascular remodeling responsible for the development of PPHN.
References
Gallot D, Coste K, Francannet C, Laurichesse H, Boda C, Ughetto S, Vanlieferinghen P, Scheye T, Vendittelli F, Labbe A, Dechelotte PJ, Sapin V, Lemery D (2006) Antenatal detection and impact on outcome of congenital diaphragmatic hernia: a 12-year experience in Auvergne, France. Eur J Obstet Gynecol Reprod Biol 125:202–205
Gucciardo L, Deprest J, Done E, Van Mieghem T, Van de Velde M, Gratacos E, Jani J, Peralta F, Nicolaides K (2008) Prediction of outcome in isolated congenital diaphragmatic hernia and its consequences for fetal therapy. Best Pract Res Clin Obstet Gynaecol 22:123–138
Pober BR (2007) Overview of epidemiology, genetics, birth defects, and chromosome abnormalities associated with CDH. Am J Med Genet C Semin Med Genet 145C:158–171
Lin AE, Pober BR, Adatia I (2007) Congenital diaphragmatic hernia and associated cardiovascular malformations: type, frequency, and impact on management. Am J Med Genet C Semin Med Genet 145C:201–216
Beppu H, Kawabata M, Hamamoto T, Chytil A, Minowa O, Noda T, Miyazono K (2000) BMP type II receptor is required for gastrulation and early development of mouse embryos. Dev Biol 221:249–258
Hogan BL (1996) Bone morphogenetic proteins in development. Curr Opin Genet Dev 6:432–438
Warburton D, Schwarz M, Tefft D, Flores-Delgado G, Anderson KD, Cardoso WV (2000) The molecular basis of lung morphogenesis. Mech Dev 92:55–81
Bellusci S, Henderson R, Winnier G, Oikawa T, Hogan BL (1996) Evidence from normal expression and targeted misexpression that bone morphogenetic protein (Bmp-4) plays a role in mouse embryonic lung morphogenesis. Development 122:1693–1702
Weaver M, Yingling JM, Dunn NR, Bellusci S, Hogan BL (1999) Bmp signaling regulates proximal–distal differentiation of endoderm in mouse lung development. Development 126:4005–4015
Alejandre-Alcazar MA, Shalamanov PD, Amarie OV, Sevilla-Perez J, Seeger W, Eickelberg O, Morty RE (2007) Temporal and spatial regulation of bone morphogenetic protein signaling in late lung development. Dev Dyn 236:2825–2835
Lu MM, Yang H, Zhang L, Shu W, Blair DG, Morrisey EE (2001) The bone morphogenic protein antagonist gremlin regulates proximal–distal patterning of the lung. Dev Dyn 222:667–680
Shi W, Zhao J, Anderson KD, Warburton D (2001) Gremlin negatively modulates BMP-4 induction of embryonic mouse lung branching morphogenesis. Am J Physiol Lung Cell Mol Physiol 280:L1030–L1039
Dewachter L, Adnot S, Guignabert C, Tu L, Marcos E, Fadel E, Humbert M, Dartevelle P, Simonneau G, Naeije R, Eddahibi S (2009) Bone morphogenetic protein signalling in heritable versus idiopathic pulmonary hypertension. Eur Respir J 34:1100–1110
Beppu H, Malhotra R, Beppu Y, Lepore JJ, Parmacek MS, Bloch KD (2009) BMP type II receptor regulates positioning of outflow tract and remodeling of atrioventricular cushion during cardiogenesis. Dev Biol 331:167–175
Wang J, Greene SB, Martin JF (2011) BMP signaling in congenital heart disease: new developments and future directions. Birth Defects Res A Clin Mol Teratol 91:441–448
de Pater E, Ciampricotti M, Priller F, Veerkamp J, Strate I, Smith K, Lagendijk AK, Schilling TF, Herzog W, Abdelilah-Seyfried S, Hammerschmidt M, Bakkers J (2012) Bmp signaling exerts opposite effects on cardiac differentiation. Circ Res 110:578–587
Rey-Parra GJ, Archer SL, Bland RD, Albertine KH, Carlton DP, Cho SC, Kirby B, Haromy A, Eaton F, Wu X, Thebaud B (2008) Blunted hypoxic pulmonary vasoconstriction in experimental neonatal chronic lung disease. Am J Respir Crit Care Med 178:399–406
Roubliova XI, Deprest JA, Biard JM, Ophalvens L, Gallot D, Jani JC, Van de Ven CP, Tibboel D, Verbeken EK (2010) Morphologic changes and methodological issues in the rabbit experimental model for diaphragmatic hernia. Histol Histopathol 25:1105–1116
Tschanz SA, Makanya AN, Haenni B, Burri PH (2003) Effects of neonatal high-dose short-term glucocorticoid treatment on the lung: a morphologic and morphometric study in the rat. Pediatr Res 53:72–80
Emery JL, Mithal A (1960) The number of alveoli in the terminal respiratory unit of man during late intrauterine life and childhood. Arch Dis Child 35:544–547
Cooney TP, Thurlbeck WM (1982) The radial alveolar count method of Emery and Mithal: a reappraisal 1—postnatal lung growth. Thorax 37:572–579
Shehata SM, Tibboel D, Sharma HS, Mooi WJ (1999) Impaired structural remodelling of pulmonary arteries in newborns with congenital diaphragmatic hernia: a histological study of 29 cases. J Pathol 189:112–118
Dewachter L, Adnot S, Fadel E, Humbert M, Maitre B, Barlier-Mur AM, Simonneau G, Hamon M, Naeije R, Eddahibi S (2006) Angiopoietin/Tie2 pathway influences smooth muscle hyperplasia in idiopathic pulmonary hypertension. Am J Respir Crit Care Med 174:1025–1033
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 72:248–254
Danesh SM, Villasenor A, Chong D, Soukup C, Cleaver O (2009) BMP and BMP receptor expression during murine organogenesis. Gene Expr Patterns 9:255–265
Southwood M, Jeffery TK, Yang X, Upton PD, Hall SM, Atkinson C, Haworth SG, Stewart S, Reynolds PN, Long L, Trembath RC, Morrell NW (2008) Regulation of bone morphogenetic protein signalling in human pulmonary vascular development. J Pathol 214:85–95
Sun J, Chen H, Chen C, Whitsett JA, Mishina Y, Bringas P Jr, Ma JC, Warburton D, Shi W (2008) Prenatal lung epithelial cell-specific abrogation of Alk3-bone morphogenetic protein signaling causes neonatal respiratory distress by disrupting distal airway formation. Am J Pathol 172:571–582
Takayasu H, Nakazawa N, Montedonico S, Puri P (2007) Down-regulation of Wnt signal pathway in nitrofen-induced hypoplastic lung. J Pediatr Surg 42:426–430
Fujiwara N, Doi T, Gosemann JH, Kutasy B, Friedmacher F, Puri P (2011) Smad1 and WIF1 genes are downregulated during saccular stage of lung development in the nitrofen rat model. Pediatr Surg Int 28:189–193
Corbett HJ, Connell MG, Fernig DG, Losty PD, Jesudason EC (2012) ANG-1 TIE-2 and BMPR signalling defects are not seen in the nitrofen model of pulmonary hypertension and congenital diaphragmatic hernia. PLoS One 7:e35364
Boucherat O, Franco-Montoya ML, Delacourt C, Martinovic J, Masse V, Elie C, Thebaud B, Benachi A, Bourbon JR (2010) Defective angiogenesis in hypoplastic human fetal lungs correlates with nitric oxide synthase deficiency that occurs despite enhanced angiopoietin-2 and VEGF. Am J Physiol Lung Cell Mol Physiol 298:L849–L856
Maniscalco WM, Watkins RH, Pryhuber GS, Bhatt A, Shea C, Huyck H (2002) Angiogenic factors and alveolar vasculature: development and alterations by injury in very premature baboons. Am J Physiol Lung Cell Mol Physiol 282:L811–L823
Du L, Sullivan CC, Chu D, Cho AJ, Kido M, Wolf PL, Yuan JX, Deutsch R, Jamieson SW, Thistlethwaite PA (2003) Signaling molecules in non-familial pulmonary hypertension. N Engl J Med 348:500–509
Bragg AD, Moses HL, Serra R (2001) Signaling to the epithelium is not sufficient to mediate all of the effects of transforming growth factor beta and bone morphogenetic protein 4 on murine embryonic lung development. Mech Dev 109:13–26
Costello CM, Cahill E, Martin F, Gaine S, McLoughlin P (2010) Role of gremlin in the lung: development and disease. Am J Respir Cell Mol Biol 42:517–523
Cahill E, Costello CM, Rowan SC, Harkin S, Howell K, Leonard MO, Southwood M, Cummins EP, Fitzpatrick SF, Taylor C, Morrell NW, Martin F, McLoughlin P (2012) Gremlin plays a key role in the pathogenesis of pulmonary hypertension. Circulation 125:920–930
Beppu H, Lei H, Bloch KD, Li E (2005) Generation of a floxed allele of the mouse BMP type II receptor gene. Genesis 41:133–137
Yang X, Castilla LH, Xu X, Li C, Gotay J, Weinstein M, Liu PP, Deng CX (1999) Angiogenesis defects and mesenchymal apoptosis in mice lacking SMAD5. Development 126:1571–1580
Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, Kalachikov S, Cayanis E, Fischer SG, Barst RJ, Hodge SE, Knowles JA (2000) Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 67:737–744
Eblaghie MC, Reedy M, Oliver T, Mishina Y, Hogan BL (2006) Evidence that autocrine signaling through Bmpr1a regulates the proliferation, survival and morphogenetic behavior of distal lung epithelial cells. Dev Biol 291:67–82
Keijzer R, Liu J, Deimling J, Tibboel D, Post M (2000) Dual-hit hypothesis explains pulmonary hypoplasia in the nitrofen model of congenital diaphragmatic hernia. Am J Pathol 156:1299–1306
Kling DE, Cavicchio AJ, Sollinger CA, Schnitzer JJ, Kinane TB, Newburg DS (2010) Nitrofen induces apoptosis independently of retinaldehyde dehydrogenase (RALDH) inhibition. Birth Defects Res B Dev Reprod Toxicol 89:223–232
Jesudason EC, Connell MG, Fernig DG, Lloyd DA, Losty PD (2000) Cell proliferation and apoptosis in experimental lung hypoplasia. J Pediatr Surg 35:129–133
Delot EC, Bahamonde ME, Zhao M, Lyons KM (2003) BMP signaling is required for septation of the outflow tract of the mammalian heart. Development 130:209–220
Kim RY, Robertson EJ, Solloway MJ (2001) Bmp6 and Bmp7 are required for cushion formation and septation in the developing mouse heart. Dev Biol 235:449–466
Roberts KE, McElroy JJ, Wong WP, Yen E, Widlitz A, Barst RJ, Knowles JA, Morse JH (2004) BMPR2 mutations in pulmonary arterial hypertension with congenital heart disease. Eur Respir J 24:371–374
Baptista MJ, Recaman M, Melo-Rocha G, Nogueira-Silva C, Roriz JM, Soares-Fernandes J, Gonzaga S, Santos M, Leite-Moreira A, Areias JC, Correia-Pinto J (2006) Myocardium expression of connexin 43, SERCA2a, and myosin heavy chain isoforms are preserved in nitrofen-induced congenital diaphragmatic hernia rat model. J Pediatr Surg 41:1532–1538
Allan DW, Greer JJ (1997) Pathogenesis of nitrofen-induced congenital diaphragmatic hernia in fetal rats. J Appl Physiol 83:338–347
Kluth D, Tenbrinck R, von Ekesparre M, Kangah R, Reich P, Brandsma A, Tibboel D, Lambrecht W (1993) The natural history of congenital diaphragmatic hernia and pulmonary hypoplasia in the embryo. J Pediatr Surg 28:456–462 Discussion 462–453
Guilbert TW, Gebb SA, Shannon JM (2000) Lung hypoplasia in the nitrofen model of congenital diaphragmatic hernia occurs early in development. Am J Physiol Lung Cell Mol Physiol 279:L1159–L1171
Acknowledgments
This work was supported by grants from the ‘Fonds de la Recherche Scientifique Médicale’ (Grant no. 3.4637.09) and the Belgian Foundation for Cardiac Surgery. MM is a doctoral fellow from the Fonds of the Medical Missionaries of Mary (Dublin, Ireland). CD and AV are doctoral fellows (‘Aspirants FNRS’) from the ‘Fonds National de la Recherche Scientifique’ (Belgium). LD was supported by a Pfizer research grant and is a postdoctoral fellow (‘Chargé de Recherche’) from the ‘Fonds National de la Recherche Scientifique’ (Belgium).
The authors are grateful for the technical assistance of Anne-Sophie Hérin and Geoffrey de Medina. They also thank Robert De Decker for helping them to gavage feed the pregnant rats.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Makanga, M., Dewachter, C., Maruyama, H. et al. Downregulated bone morphogenetic protein signaling in nitrofen-induced congenital diaphragmatic hernia. Pediatr Surg Int 29, 823–834 (2013). https://doi.org/10.1007/s00383-013-3340-6
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00383-013-3340-6