
Abstract
Purpose
The diagnosis of diffuse intrinsic pontine glioma (DIPG) is based largely on a combination of clinical and radiological findings due to the difficulty of obtaining a biopsy. An accurate evaluation of magnetic resonance imaging (MRI) scans is consequently essential. Recent analyses on the genomic landscape of DIPG revealed recurrent mutations in the H3F3A and HIST1H3B histone genes. We reviewed cases with available tumor tissue from institutional DIPG series to ascertain the consistency between their histo-molecular findings and clinical-radiological features.
Methods
We conducted a radiological and pathological central review of 22 cases enrolled in institutional DIPG trials. We performed immunohistochemical analyses to detect H3F3A/HIST1H3B K27M mutations, histone trimethylation, and EZH2 expression. Mutational analysis was performed for ACVR1, H3F3A, and HIST1H3B genes.
Results
Patients’ median age at diagnosis was 8 years, and their median overall survival was 11 months. Nineteen/22 cases (86%) showed evidence of K27M mutation on immunohistochemistry and/or mutation analysis. Histone trimethylation expression was low or lacking in these mutated cases. Sequence analysis revealed 13 cases with H3F3A and 1 case with HIST1H3B K27M mutation. There was no significant difference in EZH2 expression between the K27M mutant and wild-type DIPGs. Upon external, blinded MRI re-evaluation one lesion not consistent with DIPG showed no evidence of K27M mutation and retained histone trimethylation expression.
Conclusion
In conclusion, our study demonstrates a high frequency of histone K27M mutations in DIPG when MRI features are carefully assessed, thus confirming the consistency of imaging with biological markers in our institutional series of DIPG.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diffuse intrinsic pontine glioma (DIPG) is a malignant brainstem neoplasm of childhood for which the median survival rarely exceeds 12–24 months [1]. The tumor’s location makes it unsuitable for surgical resection, and focal radiotherapy has remained the only standard treatment [2].
In recent years, independent studies on the genomic landscape of DIPG revealed specific mutations in the H3F3A and HIST1H3B histone genes in approximately 80–90% of cases [3, 4] and recurrent mutations in the TP53, ACVR1, and PPM1D genes [5]. The 2016 WHO Classification consequently acknowledged K27M mutation in the H3F3A or HIST1H3B genes as a defining feature of a new tumor entity among diffuse midline gliomas [6], namely, DIPG. On the other hand, G34 amino acid residue mutations in the H3F3A gene have been reported in supratentorial high-grade gliomas, but not in DIPG [7].
The N-terminal tails of histones contain lysine and arginine residues that can undergo posttranslational modifications and thus regulate transcription. These changes involve acetylation, methylation, phosphorylation, ubiquitination, and SUMOylation and may prompt changes in DNA function and transcription by regulating DNA access to cellular transcriptional machinery [8]. As a prototypic example, histone H3F3A bears a lysine residue at amino acid residue position 27 that can undergo methylation and is associated with the silencing of transcription. Alterations in these processes are now known to have a central role in the pathogenesis of cancer through large-scale gene expression remodeling [9].
Since histone K27M mutations are known to alter the methylation of K27 amino acid residue, we used immunohistochemistry to investigate the expression level of trimethylation mark in tumor cells and EZH2 (a histone methyltransferase responsible for K27 methylation) [10].
In the last 20 years, we enrolled DIPG patients in a succession of institutional clinical trials, as described elsewhere [11, 12]. In these series, the diagnosis of DIPG was frequently based on a combination of clinical and radiological findings only [13]. The purpose of the present study was to seek the consistency of MRI features with the histological and biological markers in our institutional series of DIPG cases for which tissue material was available for histological and molecular reassessment.
Methods
Patients and tumor specimens
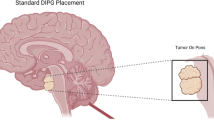
We retrieved clinical data of 45 cases for which biopsy had been performed between 1987 and 2014. Sufficient biological material could be retrieved from 12 different Italian pathology units belonging to hospitals whose neurosurgery departments sent patients to INT (Fondazione Istituto Nazionale dei Tumori, Milan) Pediatric Unit for oncological treatment. Further studies were therefore possible on 22/45 cases. Additional tumor biopsies of enrolled patients could not be analyzed due to a variety of causes, mainly missing consent or residual material. MRI findings were assessed by an external expert neuroradiologist (MW-M), blinded to any clinical data or study results, as described [12]: briefly, strict MRI eligibility criteria for verifying a DIPG were the presence of an intrinsic, pontine-based infiltrative lesion hypointense on T1-weighted sequences and hyperintense on T2-weighted sequences, involving at least 2/3 of the pons. T2-weighted sequences in all three planes were required, when available, to accurately estimate the tumor’s features. This study was approved by the review board of INT.
Immunohistochemistry
Formalin-fixed and paraffin-embedded (FFPE) material was available for immunohistochemistry in 17/22 cases. Immunohistochemical studies were performed using a Leica BondMax immunostainer, with the following antibodies: rabbit polyclonal anti-histone H3.3 K27M mutant antibody (#ABE419 Millipore, 0.5 ug/ml, 1:1000), rabbit monoclonal anti-trimethyl-histone H3 (Lys27) antibody (C36B11, Cell Signaling Tech, 1:200), and rabbit monoclonal EZH2 antibody (D2C9, Cell Signaling Tech, 1:200).
Slides were scored for H3K27M, H3K27me3, and EZH2 staining by three independent examiners, including two neuropathologists (MA, FG), who were all blinded to the tumor genotype. Samples were considered positive for H3K27 M staining if tumor cells showed strong nuclear staining; both H3.1 and H3.3K27 M mutants were recognized by anti-histone antibody, while nonneoplastic elements (such as endothelial and vascular smooth muscle cells or lymphocytes) did not stain. The H3K27me3 negative staining was considered as true negative if control blood vessels were positive for the antibody. H3K27me3 immunoreactivity and H3K27 M positivity were mutually exclusive. The EZH2 immunohistochemistry was considered positive if there was nuclear staining. EZH2 is upregulated in prostatic carcinoma, and tissue sections of prostatic adenocarcinoma were used as positive controls. Protein expression was scored as negative (score = 1), weak (2), moderate (3), or strong (4)
Mutation analysis
Genomic DNA from DIPGs was extracted from FFPE material using the QIAamp DNA FFPE Tissue kit (Qiagen). First DNA quality was assessed by polymerase chain reaction (PCR) amplification of a control 150 bp locus. Six/22 cases did not pass the quality assessment due to the age and paucity of the FFPE material. The presence of K27M mutations in histone H3F3A and HIST1H3B genes was investigated with a PCR-RFLP strategy. A 153 bp PCR-amplified fragment encompassing the mutation site was submitted to restriction endonuclease NlaIII digestion at 37 °C for 3 h. The c.211A > T point mutation creates a NlaIII restriction site, thereby dividing the PCR fragment in two (H3F3A,75 + 78 bp; HIST1H3B, 66 + 87 bp). Digested and undigested products were separated by 4% agarose gel electrophoresis and inspected with the PhotoDoc-It Imaging System (UVD, Inc.). The reference sequences are NM_002107.4 (H3F3A) and NM_003537.3 (HIST1H3B).
In addition, histone genes and ACVR1 gene coding exons were PCR-amplified using intronic primers and underwent Sanger sequencing using the 1.1 BigDye Terminator kit (ThermoFisher).
Each PCR amplification was run using TaqGold polymerase (ThermoFisher) at an annealing temperature of 55 °C, with 40 amplification cycles. The primer sequences were as follows:
Locus | Primer forward | Primer reverse | Size (bp) |
|---|---|---|---|
H3F3A exon2 | GTACAAAGCAGACTGCCCGC | CTTTGTCCCATTTTTTTCCTG | 153 |
HIST1H3B ex1 | AGACAGCTCGGAAATCCACC | TTTGGTAGCGGCGGATCTCG | 153 |
ACVR1 exon6 | CCACAGGATTTATTGGATCATTC | AACTAACATTGCATATTACCCAC | 153 |
ACVR1 exon7 | CTCCCGTGATGAGAAGTCATGG | AAAACGGAGAGAGCAAAGGC | 152 |
ACVR1 exon8 | CAGTTAGCTGCCTTCGAATAG | ATGCAACACTGTCCATTCTTC | 151 |
ACVR1 exon9 | TGGTTTAAAATCCTTCAGCAGC | ATGTAGCGCTTGGTGCCCAC | 152 |
Results
During the study period 1987–2014, a total of 137 children with brainstem gliomas were diagnosed and treated. In 45/137 (33%) patients, a surgical biopsy was performed, and we were able to retrieve sufficient biological material and consent from 22/45 cases for subsequent review of radiological, pathological, and molecular features.
Table 1 summarizes the clinical, including treatment applied and outcome, pathological, and molecular characteristics of the 22 cases on which immunohistochemistry and/or molecular analyses were feasible and MRIs were reviewed.
The patients’ median age at diagnosis was 8 years, the median duration of their symptoms at the time of diagnosis was less than 3 months, and their overall survival was 11 months. During the time span of our retrospective review, 6 different treatment protocols were applied, whose results have already been published [11, 12, 14]
On re-review, the histological diagnoses were grade 2 astrocytoma in 3 cases, grade 3 astrocytoma in 8 cases, and glioblastoma in 11 cases. All the original diagnoses had been confirmed according to WHO 2016 classification.
Radiological findings
Most MRI had been performed with a few “standard” sequences: T2 and fluid attenuated inversion recovery (FLAIR) weighted images and T1-weighted with and without contrast; very rare additional imaging sequences (e.g., diffusion, perfusion, DTI, spectroscopy) could be performed.
MRI findings, assessed by an external neuroradiologist (MW-M) blinded to any clinical data or study results, were as described [12] (Figs. 1–2). Originally [12], all but one lesion were found consistent with DIPG: the exception was of bulbar origin (ID_13, Fig. 3). All the other tumors were located primarily in the pons, encompassing at least 50% of the pons and causing its expansion. Tumors were, as it is typically described, hypo- or iso-intense on T1-weighted imaging, hyperintense on T2-weighted imaging, and relatively homogenous on fluid attenuated inversion recovery (FLAIR) sequences. They usually showed little to no contrast enhancement at initial diagnosis, but enhancement patterns varied considerably. Often, involvement of the pons encased the basilar artery.

MRI imaging of HIST1H3B-mutated DIPG case 6. Margins are unsharp; there is a patchy and multifocal contrast enhancement. No edema, necrosis, cysts, or hemorrhage are apparent

MRI imaging of K27 wild-type DIPG case 21. The postoperative MRI shows a typical DIPG with necrosis, hemorrhage and contrast enhancement. There is a postoperative injury within the pons containing cephalo-spinal fluid

MRI imaging of non-DIPG case 13. The tumor origin is nearly completely outside the pons and within medulla oblongata; margins are unsharp. The MRI performed postoperatively shows necrosis, hemorrhage, and contrast enhancement
Immunohistochemistry and molecular findings
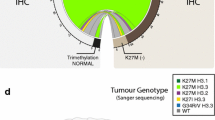
In 19/22 (86%) cases, the histone K27M mutation was identified by immunohistochemistry and/or molecular analysis (Table 1). Mutational analysis was successful in 17/22 cases and revealed the H3F3A gene K27M mutation in 13 cases and the HIST1H3B gene K27M mutation in 1 (Fig. 4 A–B). The H3F3A gene G34 amino acid residue was never found mutated in the DIPGs analyzed. Similarly, no ACVR1 mutation was detected in the case series analyzed.

DIPG histological and molecular re-evaluation.(a) H3F3A and HIST1H3B K27M mutation detected by PCR-RFLP analysis of DIPG tumor DNA (M molecular weight marker, H1 HIST1H3B gene, H3 H3F3A gene, u undigested PCR product, d NlaIII-digested PCR product). (b) Sanger sequencing electropherogram of H3F3A heterozygous mutation causing K27M substitution (c.211A > T; p.Lys27Met). (c) Immunohistochemistry of a K27M-positive DIPG case showing strong H3.3K27 M antibody nuclear positivity in tumor cells, but no staining in the nuclei of endothelial and smooth muscle cells in blood vessels. (d) Immunohistochemistry with trimethyl-histone H3(Lys27) antibody staining on the same K27M-positive DIPG sample showing an overall decline in the expression of trimethylation mark in tumor cells. (e) Immunohistochemistry for EZH2 showing a DIPG with a strong nuclear positivity in tumor cells
Among the 17/22 cases that underwent immunohistochemical analysis, positive staining with histone K27M mutant antibody was seen in 16 (Fig. 4C). Conversely, trimethyl-histone H3 (Lys27) antibody exhibited a weaker expression of trimethylation mark in tumor cells in 16/17 cases (Fig. 4D), thus showing an inverse correlation with the K27M mutant antibody (Table 1).
Combined immunohistochemistry and mutational analysis was only possible in 12 cases due to the poor quality and/or limited quantity of the material available. In all instances, these analyses produced concordant results (Table 1).
Interestingly, the one case (ID_13) originally not confirmed as DIPG on MRI reassessment (because it was of bulbar rather than pontine origin) showed no H3F3A K27M expression on immunohistochemistry; it was glioblastoma K27M wild-type on mutational analysis; and it retained a histone trimethylation expression (Table 1).
Immunohistochemistry revealed an EZH2 protein expression that was strong in 8/17 cases (Fig. 4E), moderate in 2, weak in 4, and lacking in 3. No significant differences in EZH2 expression were observed between K27M mutant and wild-type DIPGs or between trimethylation mark positive or negative DIPGs.
Discussion
Until this last decade, different treatment strategies for DIPG had failed to improve patients’ prognosis [2, 11], and radiotherapy was the only treatment definitely indicated even at relapse [15]. In recent years, a pilot protocol that combined nimotuzumab (an anti-EGFR antibody) with concomitant radiation and vinorelbine has shown a promising survival benefit and safety profile [11].
Given the limited success of mono-institutional experiences to date, some international collaborative DIPG research efforts and clinical trials have begun, and, as a first step, both the SIOPE [16] and some American institutions have established DIPG research networks that have enabled the genomic landscape of DIPG to be unraveled and the genetic alterations behind it to be identified [3,4,5].
While these recent advances are promising steps toward a better understanding of the disease’s biology and better patient care, comparisons with historical case series remain crucial in interpreting the outcome of nowadays trials – at least until the results of randomized, international clinical trials become available. Biopsies were rarely obtained in the past because this tumor is scarcely amenable to surgical resection, and little genetic information was available, so its diagnosis was based solely on a combination of clinical and radiological findings.
To test the consistency of our past institutional DIPG case series with the latest pathological and molecular data, we retrieved cases with available tumor tissue and re-reviewed their histopathological, biological, clinical, and radiological features. We identified histone K27M mutations (but not histone G34 V/R or ACVR1 mutations) in 86% of cases. It is noteworthy that the one case originally found radiologically inconsistent with a diagnosis of DIPG on external and blind re-evaluation of MRI findings concordantly showed no evidence of any K27M mutation and retained histone trimethylation expression.
While we could assess DIPG diagnosis based on typical criteria according to an expert retrospective review, we could not obtain other results on more than standard imaging sequences. Recent papers have in fact compared full description of the MRI findings at diagnosis and at relapse integrated with the molecular profiling data and clinical outcome [17, 18]. Exploring MRI sequences to compare H3 wild-type DIPG with H3 K27M mutated DIPG was beyond the scope of our work, but we consider that such a strategy will have the potential to reveal if genetic heterogeneity can correspond to specific imaging features.
EZH2 (enhancer of zeste homolog 2), also known as Enx1, is a member of the polycomb group of proteins, and it is involved in cell cycle regulation. EZH2 is expressed ubiquitously during early embryogenesis and becomes restricted to the central and peripheral nervous systems and sites of fetal hematopoiesis during later development. EZH2 represses transcription via trimethylation of histone H3 on Lys27 (H3K27), as indicated by the fact that RNAi-mediated knockdown of EZH2 resulted in a loss of H3K27 trimethylation [10]. Using immunohistochemistry, we found that EZH2 expression did not correlate with either K27M mutation or trimethylation mark, indicating that the K27M mutation in DIPG can act in a dominant negative way, suppressing trimethylation irrespective of EZH2 status.
In conclusion, the new data deriving from our past institutional case series revealed a significant consistency with recent reports on the imaging, pathological, and molecular features of DIPG. In particular, we found a strong consistency between the imaging and molecular features of DIPG, which further supports the validity and strength of our historical series for the purposes of an appropriate comparison with present and future series.
References
Hoffmann LM, van Veldhuijzen Zanten SEM, Colditz N, Baugh J, Chaney B, Hoffmann M, Lane A, Fuller C, Miles L, Hawkins C, Bartels U, Bouffet E, Goldman S, Leary S, Foreman NK, Packer R, Warren KE, Broniscer A, Kieran MW, Minturn J, Comito M, Broxson E, Shih CS, Khatua S, Chintagumpala M, Carret AS, Escorza NY, Hassall T, Ziegler DS, Gottardo N, Dholaria H, Doughman R, Benesch M, Drissi R, Nazarian J, Jabado N, Boddaert N, Varlet P, Giraud G, Castel D, Puget S, Jones C, Hulleman E, Modena P, Giagnacovo M, Antonelli M, Pietsch T, Gielen GH, DTW J, Sturm D, Pfister SM, Gerber NU, Grotzer MA, Pfaff E, von Bueren AO, Hargrave D, Solanki GA, Jadrijevic Cvrlje F, GJL K, Vandertop WP, Grill J, Bailey S, Biassoni V, Massimino M, Calmon R, Sanchez E, Bison B, Warmuth-Metz M, Leach J, Jones B, van Vuurden DG, Kramm CM, Fouladi M (2018) Clinical, radiologic, pathologic, and molecular characteristics of long-term survivors of diffuse intrinsic pontine glioma (DIPG): a collaborative report from the International and European Society for Pediatric Oncology DIPG Registries. J Clin Oncol 36:1963–1972
Donaldson SS, Laningham F, Fisher PG (2006) Advances toward an understanding of brainstem gliomas. J Clin Oncol 24:1266–1272
Wu G, Broniscer A, McEachron TA, Lu C, Paugh BS, Becksfort J, Qu C, Ding L, Huether R, Parker M, Zhang J, Gajjar A, Dyer MA, Mullighan CG, Gilbertson RJ, Mardis ER, Wilson RK, Downing JR, Ellison DW, Zhang J, Baker SJ (2012) Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 44:251–253
Gianno F, Antonelli M, Ferretti E, Massimino M, Arcella A, Giangaspero F (2018) Pediatric high-grade glioma: a heterogeneous group of neoplasms with different molecular drivers. Glioma 1:117–124
Taylor KR, Mackay A, Truffaux N, Butterfield Y, Morozova O, Philippe C, Castel D, Grasso CS, Vinci M, Carvalho D, Carcaboso AM, de Torres C, Cruz O, Mora J, Entz-Werle N, Ingram WJ, Monje M, Hargrave D, Bullock AN, Puget S, Yip S, Jones C, Grill J (2014) Recurrent activating ACVR1 mutations in diffuse intrinsic pontine glioma. Nat Genet 46:457–461
Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW (2016) The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 131:803–820
Schwartzentruber J, Korshunov A, Liu XY, Jones DT, Pfaff E, Jacob K, Sturm D, Fontebasso AM, Quang DA, Tönjes M, Hovestadt V, Albrecht S, Kool M, Nantel A, Konermann C, Lindroth A, Jäger N, Rausch T, Ryzhova M, Korbel JO, Hielscher T, Hauser P, Garami M, Klekner A, Bognar L, Ebinger M, Schuhmann MU, Scheurlen W, Pekrun A, Frühwald MC, Roggendorf W, Kramm C, Dürken M, Atkinson J, Lepage P, Montpetit A, Zakrzewska M, Zakrzewski K, Liberski PP, Dong Z, Siegel P, Kulozik AE, Zapatka M, Guha A, Malkin D, Felsberg J, Reifenberger G, von Deimling A, Ichimura K, Collins VP, Witt H, Milde T, Witt O, Zhang C, Castelo-Branco P, Lichter P, Faury D, Tabori U, Plass C, Majewski J, Pfister SM, Jabado N (2012) Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 482:226–231
Shanmugam MK, Arfuso F, Arumugam S, Chinnathambi A, Jinsong B, Warrier S, Wang LZ, Kumar AP, Ahn KS, Sethi G, Lakshmanan M (2017) Role of novel histone modifications in cancer. Oncotarget 9:11414–11426
Yuen BT, Knoepfler PS (2013) Histone H3.3 mutations: a variant path to cancer. Cancer Cell 24:567–574
Deb G, Singh AK, Gupta S (2014) EZH2: not EZHY (easy) to deal. Mol Cancer Res 12:639–653
Massimino M, Spreafico F, Biassoni V, Simonetti F, Riva D, Trecate G, Giombini S, Poggi G, Pecori E, Pignoli E, Casanova M, Ferrari A, Meazza C, Luksch R, Terenziani M, Cefalo G, Podda M, Polastri D, Clerici CA, Fossati-Bellani F, Gandola L (2008) Diffuse pontine gliomas in children: changing strategies, changing results? A mono-institutional 20-year experience. J Neuro-Oncol 87:355–361
Massimino M, Biassoni V, Miceli R, Schiavello E, Warmuth-Metz M, Modena P, Casanova M, Pecori E, Giangaspero F, Antonelli M, Buttarelli FR, Potepan P, Pollo B, Nunziata R, Spreafico F, Podda M, Anichini A, Clerici CA, Sardi I, De Cecco L, Bode U, Bach F, Gandola L (2014) Results of nimotuzumab and vinorelbine, radiation and re-irradiation for diffuse pontine glioma in childhood. J Neuro-Oncol 118:305–312
Hargrave D, Bartels U, Bouffet E (2006) Diffuse brainstem glioma in children: critical review of clinical trials. Lancet Oncol 7:241–248
Fleischhack G, Massimino M, Warmuth-Metz M, Khuhlaeva E, Janssen G, Graf N, Rutkowski S, Beilken A, Schmid I, Biassoni V, Gorelishev SK, Kramm C, Reinhard H, Schlegel PG, Kortmann RD, Reuter D, Bach F, Iznaga-Escobar NE, Bode U (2019) Nimotuzumab and radiotherapy for treatment of newly diagnosed diffuse intrinsic pontine glioma (DIPG): a phase III clinical study. J Neuro-Oncol 143(1):107–113
Janssens GO, Gandola L, Bolle S, Mandeville H, Ramos-Albiac M, van Beek K, Benghiat H, Hoeben B, Morales La Madrid A, Kortmann RD, Hargrave D, Menten J, Pecori E, Biassoni V, von Bueren AO, van Vuurden DG, Massimino M, Sturm D, Peters M, Kramm CM (2017) Survival benefit for patients with diffuse intrinsic pontine glioma (DIPG) undergoing re-irradiation at first progression: a matched-cohort analysis on behalf of the SIOP-E-HGG/DIPG working group. Eur J Cancer 73:38–47
van Veldhuijzen Zanten SE, Baugh J, Chaney B, De Jongh D, Sanchez Aliaga E, Barkhof F, Noltes J, De Wolf R, Van Dijk J, Cannarozzo A, Damen-Korbijn CM, Lieverst JA, Colditz N, Hoffmann M, Warmuth-Metz M, Bison B, Jones DT, Sturm D, Gielen GH, Jones C, Hulleman E, Calmon R, Castel D, Varlet P, Giraud G, Slavc I, Van Gool S, Jacobs S, Jadrijevic-Cvrlje F, Sumerauer D, Nysom K, Pentikainen V, Kivivuori SM, Leblond P, Entz-Werle N, von Bueren AO, Kattamis A, Hargrave DR, Hauser P, Garami M, Thorarinsdottir HK, Pears J, Gandola L, Rutkauskiene G, Janssens GO, Torsvik IK, Perek-Polnik M, Gil-da-Costa MJ, Zheludkova O, Shats L, Deak L, Kitanovski L, Cruz O, Morales La Madrid A, Holm S, Gerber N, Kebudi R, Grundy R, Lopez-Aguilar E, Zapata-Tarres M, Emmerik J, Hayden T, Bailey S, Biassoni V, Massimino M, Grill J, Vandertop WP, Kaspers GJ, Fouladi M, Kramm CM, van Vuurden DG (2017) Development of the SIOPE DIPG network, registry and imaging repository: a collaborative effort to optimize research into a rare and lethal disease. J Neuro-Oncol 132:255–266
Castel D, Philippe C, Calmon R, Le Dret L, Truffaux N, Boddaert N, Pagès M, Taylor KR, Saulnier P, Lacroix L, Mackay A, Jones C, Sainte-Rose C, Blauwblomme T, Andreiuolo F, Puget S, Grill J, Varlet P, Debily MA (2015) Histone H3F3A and HIST1H3B K27 M mutations define two subgroups of diffuse intrinsic pontine gliomas with different prognosis and phenotypes. Acta Neuropathol. 130(6):815–827
Lober RM, Cho YJ, Tang Y, Barnes PD, Edwards MS, Vogel H, Fisher PG, Monje M, Yeom KW (2014) Diffusion-weighted MRI derived apparent diffusion coefficient identifies prognostically distinct subgroups of pediatric diffuse intrinsic pontine glioma. J Neuro-Oncol 117(1):175–182
Acknowledgments
We gratefully acknowledge the unrestricted support by the following charities: Associazione Bianca Garavaglia Onlus (Busto Arsizio, I), Il Fondo di Giò (Trieste, I), Gli amici di Carola (Erba, I), Chicca Il Sole esiste per tutti (Pioltello, I), Eleonora Cocchia Vivere a Colori (Lonate Ceppino, I), and AIRC (associazione Italiana per la Ricerca sul Cancro).
Author information
Authors and Affiliations
Contributions
Experimental design: M.M., P.M, M.A. Analyses and interpretation of the data, all authors. Writing of the manuscript, all authors. Final approval of the manuscript, all authors.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Informed consent
All procedures performed were in accordance with the ethical standards of the institutional and national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Informed consent was obtained from all individual participants/legal representative included in the study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Giagnacovo, M., Antonelli, M., Biassoni, V. et al. Retrospective analysis on the consistency of MRI features with histological and molecular markers in diffuse intrinsic pontine glioma (DIPG). Childs Nerv Syst 36, 697–704 (2020). https://doi.org/10.1007/s00381-019-04463-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-019-04463-y