
Abstract
Background
Malignant rhabdoid tumor (MRT) is an aggressive tumor of infancy and childhood that rarely presents as a primary spinal or spinal cord tumor. There are only three reported cases of spinal MRT in infants.
Objective
We present a similar case in a 3-month male child who developed ultra-early recurrence, 4 weeks after complete excision. The diagnosis was confirmed by immunohistochemistry showing inactivation of the INI1 gene.
Result
Despite surgical excision and adjuvant chemoradiotherapy, these tumors have a progressive course and recurrence is a common phenomenon.
Conclusion
We believe that MRT must be considered in the differential diagnosis of the intra/paraspinal masses, especially in the infants.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Malignant rhabdoid tumors (MRT) are highly aggressive tumors of childhood, known to affect the kidney, bladder, mediastinum and paraspinal regions [1–3]. Characterized pathologically by inactivation of INI1 tumor suppressor gene on chromosome 22q11, these tumors are known for rapid proliferation and dismal outcome despite surgical excision and chemotherapy [2].
Management of such lesions when presenting in infancy is a surgical challenge, due to the low body weight of young patients and the usual large size of the lesion. Moreover, chemotherapy remains the mainstay after the surgical excision as radiotherapy cannot be given at this age. We present a case of a 3-month male infant with an intraspinal extradural rhabdoid tumor who developed ultra-early recurrence, 4 weeks after complete surgical excision. To the best of our knowledge, this is the second youngest child with this disease reported, and with the shortest time of recurrence (Table 1).
Case report
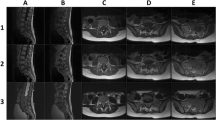
A 3-month male was brought with the history of rapidly developing weakness in the right lower limb. Examination revealed a right lower limb plegia with reduced tone. Magnetic resonance imaging (MRI) revealed an intraspinal extradural solid and cystic lesion from D12 to the S1 level that appeared isointense on T1 weighted (T1W) and T2 weighted (T2W) images (Fig. 1). There was an associated syrinx at D6 level. Intraoperatively, the extradural tumor was firm and moderately vascular, and had a well formed plane of cleavage with the dura. The operating surgeons believed that they had done a complete (gross total) resection, but no postoperative imaging was obtained. The post operative course was uneventful and the child was discharged after 5 days. There were no added deficits at the time of discharge and the monoparesis had improved marginally.

Preoperative MR images T2W sagittal (a), T1W axial (b, c), showing an intraspinal extradural solid cystic lesion extending from D12 to S1 level. The lesion is iso to hyperintense on T1W image and isointense on T2W image. In (a), a syrinx can be seen at the D6 level within the spinal cord
Histologically, the tumor was highly cellular with the cells in diffuse sheets. The individual cells were rounded to polygonal, with high nuclear cytoplasmic ratio, coarse chromatin, and prominent nucleoli (Fig. 2). Some of the cells contained an intracytoplasmic eosinophilic inclusion (rhabdoid cells). Mitotic figures were frequent. The tumor cells were infiltrating among ganglion cells of a dorsal root ganglion The tumor cells showed strong cytoplasmic immunopositivity for vimentin and focal CD99 immunopositivity. They were immunonegative for desmin, myogenin, epithelial membrane antigen (EMA), smooth muscle actin, glial fibrillary acid protein (GFAP), synaptophysin, and cytokeratin. The INI1 immunostain showed loss of nuclear INI1 immunopositivity in the tumor cells. The Ki-67 proliferation index was overall about 15 %, but, focally was as high as 30 %. Overall features were of extra-renal MRT.

a Photomicrograph showing a cellular tumor with tumor cells having high grade nuclei, prominent nucleoli, and occasional rhabdoid cells (arrow) (HE, ×400). b Tumor cells show strong cytoplasmic vimentin immunopositivity, which also highlight the rhabdoid cells (indicated by the arrows) (IP,×400). c The tumor cells show loss of nuclear INI-1 immunopositivity, while endothelial cells are immunopositive (arrows) (IP, ×200). d The tumor has a high Ki-67 proliferation index (IP, ×400)
The child was planned up for adjuvant chemotherapy, but before the same could be started, the child was brought again with a hard swelling at the operative site. A repeat MRI revealed a large recurrence extending on either side laterally with dense contrast enhancement (Fig. 3). The prognosis of the disease was explained to the parents and surgical option was offered. But they did not agree for any intervention.

Recurrent MR images T1W sagittal contrast (a), T1W axial plane (b), and T1W axial contrast (c) showing large recurrent lesion at same site with extension on either side laterally. The lesion appears isointense on T1W images and shows intense contrast enhancement
Discussion
MRT is an aggressive tumor of infancy and early childhood with a reported 2-year survival rate as low as 15 % [2]. Characterized by a loss of nuclear expression of INI1, indicating homozygous loss of SMARCB1 gene on chromosome 22q11, these tumors are known for rapid proliferation and dismal outcome despite surgical excision and adjuvant chemotherapy [2, 4]. The tumor, first described as a variant of Wilm’s tumor in the kidneys, now is known to affect the bladder, heart, mediastinum, prostate, and spine [3]. Mean age at diagnosis is 26 months; history is usually short and recurrence is common [1]. This tumor is quite distinct from the atypical teratoid/rhabdoid tumor (ATRT) that occurs within the central nervous system, has same genetic alteration as MRT and usually shows polyphenotypic differentiation towards epithelial, mesenchymal, glial, as well as neuronal differentiation [5, 6]. Our patient had an extraaxial mass showing no epithelial differentiation morphologically and had no immunopositivity for desmin, myogenin, GFAP and synaptophysin.
Spinal involvement by MRT rarely has been described in infants [1, 3]. The radiology frequently shows a large mass spanning several vertebral segments with varying degrees of contrast enhancement and necrosis [3]. The management of these lesions is a surgical challenge due to the large size of the lesion in comparison to the low body weight, and extensive spread of tumor laterally along the paraspinal muscles. The surgeon has to keep the blood loss as well as surgical duration to a minimum. This demands meticulous dissection. Since radiotherapy is not among the options at this young age, chemotherapy remains the mainstay of adjuvant therapy. Vincristine, cyclophosphamide, and actinomycin D have all been tried [2].
A thorough literature search revealed only three reports of similar spinal MRTs in infants, with recurrence seen in all of them [1, 3]. Our patient developed a large recurrence in less than 4 weeks time. The major issue, this case raises, is whether it is advisable to subject such young age children to aggressive surgical excision. The surgical insult at a 3-month age is immense and with such high proliferative potential, these tumors are fatal in most of the cases. We believe, when present in infancy, these tumors are highly lethal, and so proper prognostication is imperative. The outcome is dismal, irrespective of extent of resection achieved [2, 3]. We believe that MRT must be considered in the different diagnosis of the intra/paraspinal masses, especially in the infants. With early recurrence after surgical excision and no proven benefit of chemotherapy, a needle biopsy can be considered prior to the surgical excision. Though such lesions progress rapidly, surgical excision followed by chemotherapy seems to be a feasible option to combat the disease. The relatives need to be explained in detail the natural history of such aggressive lesions. Though the long term outcome is dismal, the presence of few survivors with MRTs, points in favor of an attempt at surgical excision.
Abbreviations
- MRT:
-
Malignant rhabdoid tumor
- MRI:
-
Magnetic resonance imaging
- ATRT:
-
Atypical teratoid rhabdoid tumor
- EMA:
-
Epithelial membrane antigen
References
Bourdeaut F, Fréneaux P, Thuille B, Bergeron C, Laurence V, Brugières L (2008) Extra-renal non-cerebral rhabdoid tumours. Pediatr Blood Cancer 51(3):363–368
Dhir A, Tekautz T, Recinos V, Murphy E, Prayson RA, Ruggieri P (2015) Lumbar spinal atypical teratoid rhabdoid tumor. J Clin Neurosci 22(12):1988–1989
Howlett DC, King AP, Jarosz JM, Stewart RA, ST A-S, Bingham JB (1997) Imaging and pathological features of primary malignant rhabdoid tumours of the brain and spine. Neuroradiology 39:719–723
Kim KH, Roberts CW (2014) Mechanisms by which SMARCB1 loss drives rhabdoid tumor growth. Cancer Genet 207(9):365–372
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds) (2007) WHO classification of tumours of the central nervous system. International Agency for Research on Cancer (IARC), Lyon
Burger PC, IT Y, Tihan T, Friedman HS, Strother DR, Kepner JL, Duffner PK, Kun LE, Perlman EJ (1998) Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a pediatric oncology group study. Am J Surg Pathol 22(9):1083–1092
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
None.
Financial disclosures
None.
Consent
The parents of the child gave informed consent for submission of the manuscript.
Rights and permissions
About this article

Cite this article
Singla, N., Kapoor, A., Chatterjee, D. et al. Ultra early recurrence in giant congenital malignant rhabdoid tumor of spine. Childs Nerv Syst 32, 2471–2474 (2016). https://doi.org/10.1007/s00381-016-3178-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-016-3178-z