
Abstract
Background
Extradural malignant rhabdoid tumors of the spine are highly malignant and invasive tumors (WHO grade IV) with poor prognosis, most frequently occurring in young children before 2 years of age. Pain and motor deficit are the most common presenting signs.
Case description
We report a case of a 2-year-old girl presenting with axial ataxia and paraparesis related to an extradural malignant rhabdoid tumor causing posterior thoracic spinal cord compression (D3–D6). She underwent two near-total removal of the tumor, adjuvant chemotherapy according to the Eu-Rhab protocol and proton beam therapy. She then developed multiple cranial nerve paresis (meningeal carcinomatosis) after 4 cycles of chemotherapy and died at 4.32 months of follow-up.
Discussion and conclusion
The role of the PET scan was essential to guide us to remove a residue, while two concomitant spinal MRIs were considered negative. We reviewed the 16 cases reported in the literature. Multiple surgeries and radiotherapy seem to be correlated with longer survival. No child younger than 2 years old had a documented survival higher than 4.32 months.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Malignant rhabdoid tumors (MRTs) are highly aggressive embryonal tumors (WHO grade IV) that usually arise in young children (median age at onset, 11–18 months). The incidence of MRT is very low, estimated at 0.06 per 100,000 [1]. The three presentations of MRT (location-based) are MRT of the kidney (MRTK), MRT of the central nervous system (atypical teratoid rhabdoid tumor—ATRT) and extracranial, extrarenal rhabdoid tumor (EERT) [2]. Nevertheless, the literature is unclear for lesions occurring outside the dura but inside the spinal canal, and they are classified as EERT or extradural primary spinal ATRT. Even though ATRT is the most common CNS malignancy in children younger than 6 months of age and accounts for 4.4% of CNS tumors in the age group 0–5 years [3], primary spinal cases are rare, with only 49 pediatric cases (9 purely extradural) documented in the English-language literature according to Li et al. [4].
Historical background
The low median age of onset of this cancer may be explained by the paucity of mutations found and needed for its development. According to Lee et al. [5], the mean mutation rate is 0.19 mutations per Mb, the lowest of all high-grade cancers sequenced to date. In most of these tumors, the only genes showing recurrent inactivation are SMARCB1 or, in rare cases, SMARCA4, two genes encoding subunit members of the BAF chromatin-remodeling complex that are essential for the organization of chromatin and activation of most promoters [6, 7].
Approximately 20 to 35% of patients presenting with MRT have heterozygous germline mutations in one of those two genes, defining rhabdoid predisposition syndrome 1 (for monoallelic SMARCB1 mutation) or 2 (for monoallelic SMARCA4 mutation) [7, 8]. These predisposition syndromes lead to familial cases and earlier onset of rhabdoid tumors, in agreement with the two-hit model (median age of onset of 5.5 months in children with RSP1) [7].
We reviewed the literature regarding extradural malignant rhabdoid tumors and found 16 other similar cases of tumors that were nonmetastatic at diagnosis [2, 3, 9,10,11,12,13,14,15,16,17,18,19]. We summarize the relevant information in Table 1.
Clinical presentation
The most common symptom is pain, followed by motor deficits. The median duration of symptoms before diagnosis is 6 weeks. There is no significant gender difference (46% male and 54% female). The mean age at diagnosis is 4.29 years, and the median age is 1.75 (Table 1).
Diagnosis
The radiological appearance of the lesion compared to the spinal cord is often hypo/iso T1 and iso/hyper T2, with heterogeneous enhancement after gadolinium injection. Bony involvement is frequent, with foraminal enlargement or vertebral body osteolysis. All the lesions compressed the neural elements at diagnosis, most of the time anteriorly (70%). The cervical spine is the most impacted segment (50%) (Table 1).
MRT are solid tumors, composed of poorly differentiated (rhabdoid) cells with prominent nucleoli. The diagnosis is confirmed by DNA methylation profiling.
Management
There is no clear consensus about the management of these lesions. Most frequently therapeutic management consists of surgery followed by chemotherapy and radiotherapy (38%) or surgery followed by chemotherapy alone (38%). Surgery alone is performed in 13% of the cases, and chemotherapy or radiotherapy alone in 6%. Outcomes are detailed in Table 1.
Prognosis and outcomes
Prognosis is poor, with survival rates between 0.4 and 42 months (mean of 12 months) [20]. Unfavorable prognostic factors are age below 2 years, metastatic disease at diagnosis, non-R0 tumoral resection, and delayed initiation of radiation therapy [3, 6]. All patients with documented survival of more than 1 year (four) were at least 2.25 years of age at diagnosis and underwent at least 2 surgeries and radiotherapy. The location of the lesion was documented in three out of these four cases and was localized anteriorly in the cervical spine between C0 and C4 for all 4 patients. Two patients [3, 17] had a survival superior to 40 months. They were 7 and 13 years old at diagnosis. Treatment included gross-total resection of the lesion. They both received several lines of chemotherapy (including vincristine and etoposide) and radiotherapy. The second child [17] also benefitted from the implantation of seven 125I radioactive seeds into the tumoral area. The first child [3] had extramedullary metastasis at the L1 level 27 months after the original surgery. He underwent a laminectomy from T-12 to L-1, and the tumor was removed. He then underwent high-dose chemotherapy and a stem cell rescue clinical trial following resection but died of disseminated disease 42 months after the first surgery. The second patient [17] had a follow-up of 40 months, with no recurrence or new lesion onset at the last follow-up.
Exemplary case description
A 2-year-old girl was admitted to the emergency department after a 3-day history of intense discomfort, irritability with inconsolable crying upon touching her back and chest and sleep difficulties. She also developed a brutal onset of axial ataxia and paraparesis the day before admission. Blood tests were normal. Lumbar puncture showed hyperproteinorrhachia without other anomalies.
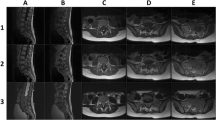
Spine MRI showed a well-defined extradural mass located in D3–D6, pushing the dural sac and the spinal cord forward, associated with altered spinal cord signal on T2-weighted images (T2 hypersignal). The tumor presented an intermediate T2, STIR, and T1 postenhancement with a well-defined, hypo T2 capsule. The lesion extended toward the D4–D5 and D5–D6 foramina, predominantly on the right, with enlarged foramina suggesting a relatively slow process. The magnetic susceptibility sequence showed no hematic residue. The diffusion sequence demonstrated a clear and homogeneous restriction, with a drop in signal on the ADC map. The rest of the spine and the head MRI were normal (Fig. 1).

MRI of the cervical and thoracic spine (day 0). a Sagittal T1 image, b sagittal T2 image, and c axial T2 image
The patient was brought to the operative room the same day for medullar decompression and tumor resection. We found a hemorrhagic, soft yellowish extradural tumor. There was no dural attachment or dural alteration. The lesion could easily be removed from the dura using a dissector. Foraminal resection was performed using an ultrasonic aspirator (CUSA®) (Fig. 2). The resection was considered complete intraoperatively and on the first look of the postoperative MRI.

Perioperative pictures of the lesion. Upper part of the lesion on the left side of the pictures a before tumor resection, b during tumor resection, and c at the end of tumor resection
Paraparesis remained severe (Medical Research Council (MRC) 2) in the immediate postoperative period but improved quickly thereafter (MRC 3–4 at discharge on day 6).
Histological examination revealed a solid tumor surrounded by adipose tissue. The neoplasm was composed of sheets of poorly differentiated cells with prominent nucleoli and scant cytoplasm and exhibited frequent mitotic and apoptotic figures. Some areas showed a myxoid background, and foci of necrosis were present as well (Fig. 3a).

Histopathological and immunohistochemical findings. H&E staining revealed poorly differentiated cells with prominent nucleoli, scant cytoplasm, and frequent mitotic figures (arrows) (a). The MIB-1/Ki67 labeling index was up to 80% of tumor cells (b). INI-1 loss of nuclear staining in tumoral cells (c)
The immunohistochemical analysis showed partial positivity for cytokeratin AE1/AE3, EMA, and SALL4, while GFAP, S100, and synaptophysin were negative. The MIB-1/Ki-67 labeling index was 80%. INI-1 nuclear staining was lost in the tumoral cells (Fig. 3b, c).
The diagnosis of extrarenal rhabdoid tumor was thus established and confirmed by DNA methylation profiling.
Analysis of the SMARCB1 gene in the tumor found a homozygous deletion of SMARCB1 in the methylation profiling.
To exclude extraspinal metastasis of MRTK, we performed a full-body PET-CT scan (FDG) (day 10) and MRI (day 12) (Fig. 4a, b). It revealed a probable nodule next to the right D5–D6 foramina embedded in the postsurgical inflammatory tissue. On second look, the residue was visible on the postoperative MRI (day 5) (Fig. 4c).

Full body PET-CT (day 10) a Axial view of the D5–D6 level, b sagittal view of the D5-D6 level, and c postoperative MRI of the D5–D6 level (day 5)
The child underwent a second surgery, and resection of the residue was considered again complete (day 26) (Fig. 5).

Operating view of the tumor inside the right D5–D6 foramen before (a) and after (b) its opening
Adjuvant chemotherapy was started on day 36, according to the Eu-Rhab protocol [20], consisting of sequential chemotherapies starting on day 36 (DOX (doxorubicin), ICE (ifosfamide, carboplatinum, and etoposide), and VCA (vincristine, cyclophosphamide, and actinomycin-D)) and radiotherapy. The patient underwent proton beam therapy (50.4 Gy in 28 fractions of 1.8 Gy, 4–5 times a week) starting on day 58.
A third spinal MRI was performed before proton beam therapy (day 44) (Fig. 6). Among the important postsurgical inflammatory changes, a nodular image at the height of the D5 vertebral body was found. It was located in the right paravertebral region, extending between the subpleural space and the D5–D6 conjugation foramen. It was not visible on the previous MRI scan (day 12), but intercostal extension was retrospectively visible on the PET scan (day 10).

MRI of the thoracic spine (day 44). a Axial T1+gadolinium, b coronal T1+gadolinium, and c PET- CT of the thoracic spine (day 10)
Unfortunately, the patient developed multiple cranial nerve paresis after 4 cycles of chemotherapy. MRI (day 121) showed meningeal carcinomatosis with multiple intracranial metastases and within the spinal canal. The child died on day 134.
Conclusion
Extradural malignant rhabdoid tumors of the spine are highly malignant and invasive tumors with poor prognosis, most frequently occurring in young children before 2 years of age. The most common symptoms are pain and motor deficits. Diagnosis could be suggested when imaging depicts a large extradural mass that extends through the intervertebral foramina and invades the paraspinal soft tissues or vertebral bones in a young patient.
Due to its MRI signal, recurrence does not always appear clear on MRI, and FDG PET scans can deliver precious assessment information about the location and size of the residue or tumoral progression. Multiple surgeries and radiotherapy seem to be correlated with longer survival.
Availability of data and material
All medical data are available and recorded in a secure computerized medical file.
Code availability
Not applicable.
References
Xie S, Yang J, Ma Y, Li K, Dong K, Yao W (2022) Analysis on diagnosis and treatments of 16 cases of extracranial malignant rhabdoid tumor in children. Transl Cancer Res 11(4):629–638
Uwineza A, Gill H, Buckley P, Owens C, Capra M, O’Sullivan C, McDermott M, Brett F, Farrell M, Pears J, O’Sullivan MJ (2014) Rhabdoid tumor: the Irish experience 1986–2013. Cancer Genet 207(9):398–402
Heuer GG, Kiefer H, Judkins AR, Belasco J, Biegel JA, Jackson EM et al (2010) Surgical treatment of a clival-C2 atypical teratoid/rhabdoid tumor. J Neurosurg Pediatr 5:75–79
Li D, Heiferman DM, Syed HR, Santos JG, Bowman RM, DiPatri AJ Jr, Tomita T, Wadhwani NR, Alden TD (2019) Pediatric primary spinal atypical teratoid rhabdoid tumor: a case series and review of the literature. J Neurosurg Pediatr 24(3):267–283
Lee RS, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, Lawrence MS, Auclair D, Mora J, Golub TR et al (2012) A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Investig 122:2983–2988
Hasselblatt M, Gesk S, Oyen F, Rossi S, Viscardi E, Giangaspero F, Giannini C, Judkins AR, Frühwald MC, Obser T, Schneppenheim R, Siebert R, Paulus W (2011) Nonsense mutation and inactivation of SMARCA4 (BRG1) in an atypical teratoid/rhabdoid tumor showing retained SMARCB1 (INI1) expression. Am J Surg Pathol 35(6):933–935
Schneppenheim R, Frühwald MC, Gesk S, Hasselblatt M, Jeibmann A, Kordes U, Kreuz M, Leuschner I, Subero JIM, Obser T et al (2010) Germline nonsense mutation and somatic inactivation of SMARCA4/BRG1 in a family with rhabdoid tumor predisposition syndrome. Am J Hum Genet 86:279–284
Kenny C, O’Meara E, Ulaş M, Hokamp K, O’Sullivan MJ (2021) Global chromatin changes resulting from single-gene inactivation-the role of SMARCB1 in malignant rhabdoid tumor. Cancers (Basel) 13(11):2561
Agrawal A, Bhake A, Cincu R (2009) Giant lumbar paraspinal atypical teratoid/rhabdoid tumor in a child. J Cancer Res Ther 5:318–320
Bourdeaut F, Fréneaux P, Thuille B, Bergeron C, Laurence V, Brugières L (2008) Extra-renal non-cerebral rhabdoid tumours. Pediatr Blood Cancer 51(3):363–368
Dobbs MD, Correa H, Schwartz HS, Kan JH (2011) Extrarenal rhabdoid tumor mimicking a sacral peripheral nerve sheath tumor. Skeletal Radiol 40:1363–1368
Horie H, Etoh T, Maie M (1992) Cytogenetic characteristics of a malignant rhabdoid tumor arising from the paravertebral region. A case report. Acta Pathol Jpn 42:460–465
Mahmood MN, Salama ME, Shah VV (2003) Pathologic quiz case: an 11-year-old boy with cervical lymphadenopathy. Metastatic malignant extrarenal rhabdoid tumor. Arch Pathol Lab Med 127:e361–e362
Nishimoto T, Nomura S, Fukano R, Kimura T, Ikeda E, Suzuki M (2018) A primary extradural malignant rhabdoid tumor at the craniovertebral junction in a 3-year-old boy. Childs Nerv Syst 34:367–371
Singla N, Kapoor A, Chatterjee D, Radotra BD (2016) Ultra early recurrence in giant congenital malignant rhabdoid tumor of spine. Childs Nerv Syst 32:2471–2474
Tamiya T, Nakashima H, Ono Y, Kawada S, Hamazaki S, Furuta T et al (2000) Spinal atypical teratoid/rhabdoid tumor in an infant. Pediatr Neurosurg 32:145–149
Tang Y, Li S, Qu J, Zhou Y, Xiao J (2015) Malignant rhabdoid tumor with cervical vertebra involvement in a teenage child: case report and review of the literature. Pediatr Neurosurg 50:173–178
Tsitsopoulos PP, Marinos K, Chochliourou E, Theologou M, Nikolaidou C, Sdougka M, Tsonidis CA (2020) Infantile atypical teratoid rhabdoid tumor of the spine presenting with acute hydrocephalus. Pediatr Neurosurg 55(5):313–318
Xin X, Zhu B, Shen J, Tian C, Fan X, Liu BR (2014) A primary spinal extradural atypical teratoid/rhabdoid tumor of the cervical spine with bony involvement. J Child Neurol 29(5):670–673
Bartelheim K, Nemes K, Seeringer A, Kerl K, Buechner J, Boos J, Graf N, Dürken M, Gerss J, Hasselblatt M, Kortmann RD, Teichert von Luettichau I, Nagel I, Nygaard R, Oyen F, Quiroga E, Schlegel PG, Schmid I, Schneppenheim R, Siebert R, Solano-Paez P, Timmermann B, Warmuth-Metz M, Frühwald MC (2016) Improved 6-year overall survival in AT/RT - results of the registry study Rhabdoid 2007. Cancer Med 5(8):1765–1775
Funding
No financial support was received for this work.
Author information
Authors and Affiliations
Contributions
EV: conception of the draft, conception of the figures and tables, clinical management of the patient. HR: substantial contributions to the conception of the work, clinical management of the patient. VJ: substantial contributions to the conception and design of the work, clinical management of the patient. LD: substantial contributions to the conception and design of the work, conception of the figures. AVD: conception of the draft, substantial contributions to the conception and design of the work, clinical management of the patient. CR: substantial contributions to the conception and design of the work.
Corresponding author
Ethics declarations
Ethics approval
The patient’s family has consented to the submission of the case report for submission to the journal.
Consent to participate
The patient’s family has consented to the submission of the case report for submission to the journal.
Consent for publication
The patient’s family has consented to the submission of the case report for submission to the journal.
Conflict of interest
The authors declare no competing interests.
Disclaimer
The manuscript is an original contribution, not submitted simultaneously in another journal or published elsewhere, partially or in full in any form or language. Results are presented clearly, honestly, and without fabrication, falsification or inappropriate data manipulation (including image based manipulation). Authors adhere to discipline-specific rules for acquiring, selecting and processing data.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Vigneul, E., Rooijakkers, H., Joris, V. et al. Extradural malignant rhabdoid tumor of the spine in children: A case-based review. Childs Nerv Syst 40, 979–986 (2024). https://doi.org/10.1007/s00381-023-06224-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-023-06224-4