
Abstract
Purpose
Desmoplastic infantile gliomas (DIG) are rare tumors that occur in infants aged between 1 and 24 months. The tumor in general has a favorable prognosis after surgical resection. There are no treatment algorithms, however, for patients with multiple intracranial and intraspinal presentations.
Case report
In an 11-month-old girl with a history of nystagmus, magnetic resonance imaging (MRI) demonstrated contrast-enhancing lesions in the suprasellar region, the cerebellar vermis, and the spinal axis. The tumor in the cerebellar vermis was removed via a suboccipital midline approach. The histological examination revealed a desmoplastic infantile astrocytoma (DIA) WHO grade I. Postoperatively, it was decided to adopt a wait-and-see strategy. Further development, up to 16 months after surgery, was unremarkable. Follow-up MRI showed no recurrence of the posterior fossa tumor, mild progress of the suprasellar tumor, and significant regression of the spinal tumors.
Conclusion
DIA is a rare mostly benign brain tumor found in infants. The final diagnosis always relies on histology. Surgical resection is the recommended therapy for symptomatic tumors; however, more experience is needed to develop treatment recommendations for multiple-site tumors.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Desmoplastic infantile glioma, described first in 1984 by Taratuto et al. [18], is a rare tumor which presents most often as a large hemispheric mass [9]. These tumors usually occur in infants aged between 1 and 24 months with a male/female ratio of 1.7/1.0 [4]. Radiologically, they may manifest as large cystic and solid well-defined supratentorial lesions that involve the cerebral cortex and leptomeninges, attached to the dura mater [19]. Two histologic forms, which were integrated in the same tumor class according to the World Health Organization classification, have been described: desmoplastic infantile astrocytoma (DIA) and desmoplastic infantile ganglioglioma (DIG) [4, 19]. The presence of a neuronal component in DIG is the only difference between both subtypes [4]. When surgical resection of the tumor is feasible, a generally favorable prognosis may be expected. The management of patients with multiple intracranial and intraspinal tumors, however, remains to be defined because of limited experience [2, 4, 5]. The current report describes the multiple occurrence of DIG in the suprasellar region and the cerebellar vermis with additional spinal tumors in an infant and its practical management.
Case report
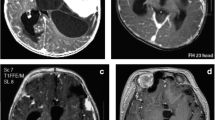
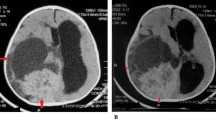
An 11-month-old girl, who was born at term after uncomplicated pregnancy and delivery, was referred to our clinic with a history of nystagmus for 2 weeks. On admission, examination revealed no other neurological deficits. Her past medical history was unremarkable. Physical and blood serum examinations revealed no abnormalities. T1-weighted gadolinium-enhanced magnetic resonance imaging (MRI) showed a contrast-enhancing mass in the suprasellar region. Another enhancing lesion with cystic components was seen in the cerebellar vermis extending to the lateral recess (Fig. 1). MRI of the spine showed multiple spinal tumors in addition (Fig. 2).

Axial and coronal postgadolinium T1-weighted MRI revealed a large enhancing mass in the suprasellar region (a, b) and another enhancing tumor in the lower cerebellar vermis extending to the lateral recess (c, d)

Sagittal postgadolinium T1-weighted MRI showed multiple spinal tumors in addition (white arrows)
The tumor in the cerebellar vermis was resected via a midline osteoplastic suboccipital craniotomy to obtain a histopathological diagnosis and to avert the development of occlusive hydrocephalus.
The histological examination revealed a desmoplastic glial tumor with moderate cellularity and low pleomorphism. It was predominately composed of spindle-shaped cells arranged in fascicles or in a storiform pattern (Fig. 3a) forming a prominent network of argyrophilic reticulin fibers, which surrounded nearly each tumor cell (Fig. 3b). EvG staining demonstrated a collagen-rich matrix. Other tumor cells with a round to oval shape showed a clear to faint eosinophilic matrix (Fig. 3c). These cells were partially arranged in small groups forming nests of tumor cells. Very rarely, small foci of poorly differentiated cells with round nuclei with dense chromatin showed up between astrocytic tumor regions (Fig. 3c). Other areas exhibited a nodular pattern with a more loosened texture with microcystic structures and partially biphasically oriented astrocytic tumor cells (Fig. 3d). In sharply demarcated regions, the dense network of reticulin fibers was missing (Fig. 3d). Neither a poorly differentiated primitive small blue cell component nor a clear ganglion cell component was observed. The predominately oval-shaped nuclei had loose chromatin. Prominent nucleoli were seen only occasionally, and mitotic figures were absent. In certain areas of the tumor tissue, a dense network of unsuspicious vessels was seen. Necrotic areas were lacking. The proliferation rate was around 2 %, and many tumor cells showed expression of GFAP (Fig. 3e). MAP2, Vimentin, and S100 were expressed by nearly all cells. Synaptophysin- and chromogranin-positive ganglion-like elements were not observed. Predominately, not only the biphasic astrocytic tumor cells of the nodular regions, but also the tumor cells of other areas, expressed mutated BRAF V600E protein (Fig. 3f). The desmoplastic tumor component, however, showed no clear expression of this epitope. After DNA extraction and pyrosequencing, an underlying BRAF V600E mutation was confirmed.

Desmoplastic component of spindle-shaped cells arranged in fascicles or in a storiform pattern (a) with a dense network of reticulin fibers (d) (plus); other tumor cells showed a clear to faint eosinophilic cytoplasm (b) (arrow head); small foci of poorly differentiated neuroepithelial cells with round nuclei and a dense chromatin (b) (arrow); sharply demarcated cortical regions composed of partially biphasic astrocytic tumor cells (c) (star) without reticulin fibers (d) (star); predominately, the biphasic astrocytic tumor cells of the cortical component expressed mutated BRAF V600E protein (e); many of the astrocytic tumor cells expressed GFAP (f)
After the surgery, there were no new neurological deficits. The findings and possible treatment points were discussed in our interdisciplinary neuro-oncology board, and a wait-and-see strategy was favored with regard to both the clinical status and age.
Further development was unremarkable, and the patient was doing well for 16 months after surgery without tumor recurrence in the posterior fossa. Mild enlargement of the tumor mass in the suprasellar region was detected, while there was a regression of the multiple spinal tumors. Clinically, the patient was stable without neurological or endocrinological symptoms.
Discussion
According to the WHO classification of brain tumors, desmoplastic infantile gliomas are composed of three distinctive components: (i) a main desmoplastic leptomeningeal component, (ii) a poorly differentiated neuroepithelial component, and (iii) a cortical component [13]. We categorized the spindle-shaped tumor areas in our patient as a desmoplastic component, and the poorly differentiated cells as a neuroepithelial component, and the nodular areas without reticulin fibers as a cortical component and, therefore, diagnosed a DIA WHO grade I.
DIA/DIG usually grows slowly and appears to have a good prognosis after surgical resection alone [15]. Increased intracranial pressure (dysphoria, vomiting), bulging fontanelle, abnormally increased head circumference, seizures, and sensorimotor deficits are the most frequent clinical manifestations of DIA/DIG [7]. Sometimes intracranial DIA/DIGs have both solid and cystic components. The cystic component (uni- or multilocular) seems to be located more deeply inside of the lesion, whereas its solid part tends to be more peripherally [15, 19].
The relation of DIA/DIG to other brain tumors remains unclear, but the morphological overlap of certain elements with features of either pilocytic astrocytoma or ganglioglioma is remarkable. On the other hand, because of their primitive small cell components, DIA/DIG has been discussed also to represent an embryonal neoplasm with progressive maturation [13]. Only recently, BRAF V600E mutations were described in a fraction of these tumors [6, 10]. BRAF V600E mutations occur in 60 % of gangliogliomas [11] and in 5 % of pilocytic astrocytomas. However, also more than 60 % of pilocytic astrocytomas harbor BRAF:KIAA1549 duplications and deletions [8]. The detection of BRAF V600E mutations in DIA/DIG supports the notion that these tumors may indeed be more related to gangliogliomas and pilocytic astrocytomas regarding their origin. Expression of mutated BRAF V600E protein was predominately observed in the cortical tumor component and not in the desmoplastic areas [10]. This observation may indicate that the true neoplastic tumor component of a DIA/DIG is solely reflected by those smaller nodular areas without reticulin fibers, whereas the desmoplastic areas that dominate the tumor may be non-neoplastic, reactive areas.
DIA/DIG has been shown to occur at multiple sites in few instances [4, 5, 17, 20]. Such biological behavior was accompanied by anaplastic transformation and short survival in one instance [5]. In our patient, the tissue that became subject for neuropathological evaluation showed no signs of malignancy, and we did not initiate adjuvant treatment. It is conceivable that patients with progressive DIA/DIG with BRAF V600E mutations that extend towards a systemic disease might have a benefit from selective BRAF V600E inhibitors regarding their effect in patients with metastatic melanomas [3] and other pediatric brain tumors [1, 14, 16].
Table 1 summarizes reported cases of multiple-site intracranial and intraspinal DIA/DIGs at presentation. Setty et al. [17] reported a 4-month-old boy with nystagmus and macrocephaly. He had a large tumor in the suprasellar and hypothalamic region, as well as two smaller tumors in the posterior fossa and in the spinal canal. A biopsy of the suprasellar mass was performed which confirmed the diagnosis of DIA. The authors postulated that the smaller tumors were metastases from the large suprasellar primary astrocytoma. De Munnynck et al. [5] reported a 2-year-old girl with a large tumor predominately in the right hemisphere but with extension over the midline and a separate nodular lesion in the hypothalamic region as well as diffuse pial and dural-enhancing lesions, particularly in the thoracic and lumbar region. The large tumor was removed. After 5 weeks, the girl presented with paraplegia, incontinence, and multiple cranial nerve deficits. Brain MRI 1 month after surgery demonstrated rapid growth of the hypothalamic mass as well as of the pial and of the ependymal tumors. Chemotherapy with vincristine and carboplatinum was started 12 months after surgery; however, it did not prevent death secondary to tumor progression [5]. Darwish et al. presented a 3-month-old boy with a multiloculated cystic right hemispheric tumor with a solid enhancing component and multiple spinal tumors [4]. After removal of the peripheral enhancing component of the tumor, a drain was placed in the cystic component. An MRI scan of the craniospinal axis 1 month later showed further increase in the size and number of the spinal tumors. Chemotherapy with vincristine and carboplatin according to the Low Grade Glioma Protocol of the International Society of Pediatric Oncology was installed. With this regimen, a further increase in the number and size of the leptomeningeal metastases was noted on repeated MRI. Therefore, medication was switched to oral temozolomide. Eight months later, he was alive and well without neurological deficits [4].
It is difficult in infantile cerebral neoplasms to predict tumor histology and grading on the basis of the preoperative imaging, and surgical resection or biopsy to establish a diagnosis has been recommended [12, 21]. Surgical resection has also been favored for treatment of DIA, and good quality of life can be achieved on long-term follow-up in patients with a single mass [7, 21]. In cases with multiple intracranial lesions, surgical resection of the symptomatic mass and follow-up of the other non-symptomatic lesions in this classically benign entity might be an option. Reoperating on stable residual masses or resection of non-symptomatic lesions can be questioned in patients with benign entities [20]. Because of the small number of reported cases of multiple DIA, the role of adjuvant therapy such as radiation and chemotherapy with selective BRAF inhibitors like vemurafenib for these tumors still needs to be clarified. Furthermore, it is unclear if such cases with multiple DIA are due to metastatic spreading or if such tumors are growing independent from each other due to an inherited genetic disorder.
In conclusion, DIA/DIG is a rare, mostly benign brain tumor found in infants. The final diagnosis always relies on histology. Multifocal appearance which may be due to metastasis to the spinal cord has been reported occasionally. Surgical resection is the recommended therapy for DIA/DIG, while more experience is needed to develop treatment recommendations for multiple-site tumors.
References
Bautista F, Paci A, Minard-Colin V, Dufour C, Grill J, Lacroix L, Varlet P, Valteau-Couanet D, Geoerger B (2014) Vemurafenib in pediatric patients with BRAFV600E mutated high-grade gliomas. Pediatr Blood Cancer 61:1101–1103
Beppu T, Sato Y, Uesugi N, Kuzu Y, Ogasawara K, Ogawa A (2008) Desmoplastic infantile astrocytoma and characteristics of the accompanying cyst. Case report. J Neurosurg Pediatr 1:148–151
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, O’Day SJ, Sosman JA, Kirkwood JM, Eggermont AM, Dreno B, Nolop K, Li J, Nelson B, Hou J, Lee RJ, Flaherty KT, McArthur GA (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364:2507–2516
Darwish B, Arbuckle S, Kellie S, Besser M, Chaseling R (2007) Desmoplastic infantile ganglioglioma/astrocytoma with cerebrospinal metastasis. J Clin Neurosci 14:498–501
De Munnynck K, Van Gool S, Van Calenbergh F, Demaerel P, Uyttebroeck A, Buyse G, Sciot R (2002) Desmoplastic infantile ganglioglioma: a potentially malignant tumor? Am J Surg Pathol 26:1515–1522
Dougherty MJ, Santi M, Brose MS, Ma C, Resnick AC, Sievert AJ, Storm PB, Biegel JA (2010) Activating mutations in BRAF characterize a spectrum of pediatric low-grade gliomas. Neuro Oncol 12:621–630
Gu S, Bao N, Yin MZ (2010) Combined fontanelle puncture and surgical operation in treatment of desmoplastic infantile astrocytoma: case report and a review of the literature. J Child Neurol 25:216–221
Jones DT, Hutter B, Jäger N, Korshunov A, Kool M, Warnatz HJ, Zichner T, Lambert SR, Ryzhova M, Quang DA, Fontebasso AM, Stütz AM et al (2013) Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 45:927–932
Kato M, Yano H, Okumura A, Shinoda J, Sakai N, Shimokawa K (2004) A non-infantile case of desmoplastic infantile astrocytoma. Childs Nerv Syst 20:499–501
Koelsche C, Wohrer A, Jeibmann A, Schittenhelm J, Schindler G, Preusser M, Lasitschka F, von Deimling A, Capper D (2013) Mutant BRAF V600E protein in ganglioglioma is predominantly expressed by neuronal tumor cells. Acta Neuropathol 125:891–900
Koelsche C, Sahm F, Paulus W, Mittelbronn M, Giangaspero F, Antonelli M, Meyer J, Lasitschka F, von Deimling A, Reuss D (2014) BRAF V600E expression and distribution in desmoplastic infantile astrocytoma/ganglioglioma. Neuropathol Appl Neurobiol 40:337–344
Lang SS, Beslow LA, Gabel B, Judkins AR, Fischer MJ, Sutton LN, Storm PB, Heuer GG (2012) Surgical treatment of brain tumors in infants younger than six months of age and review of the literature. World Neurosurg 78:137–144
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109
Nicolaides TP, Li H, Solomon DA, Hariono S, Hashizume R, Barkovich K, Baker SJ, Paugh BS, Jones C, Forshew T, Hindley GF, Hodgson JG, Kim JS, Rowitch DH, Weiss WA, Waldman TA, James CD (2011) Targeted therapy for BRAFV600E malignant astrocytoma. Clin Cancer Res 17:7595–7604
Rasalkar DD, Paunipagar BK, Ng A (2012) Primary spinal cord desmoplastic astrocytoma in an adolescent: a rare tumour at rare site and rare age. Hong Kong Med J 18:253–255
Rush S, Foreman N, Liu A (2013) Brainstem ganglioglioma successfully treated with vemurafenib. J Clin Oncol 31:e159–e160
Setty SN, Miller DC, Camras L et al (1997) Desmoplastic infantile astrocytoma with metastases at presentation. Mod Pathol 10:945–951
Taratuto AL, Monges J, Lylyk P, Leiguarda R (1984) Superficial cerebral astrocytoma attached to dura: report of six cases in infants. Cancer 54:2505–2512
Trehan G, Bruge H, Vinchon M, Khalil C, Ruchoux MM, Dhellemmes P, Ares GS (2004) MR imaging in the diagnosis of desmoplastic infantile tumor: retrospective study of six cases. AJNR Am J Neuroradiol 25:1028–1033
Uro-Coste E, Ssi-Yan-Kai G, Guilbeau-Frugier C, Boetto S, Bertozzi AI, Sevely A, Lolmede K, Delisle MB (2010) Desmoplastic infantile astrocytoma with benign histological phenotype and multiple intracranial localizations at presentation. J Neurooncol 98:143–149
Vandenberg SR (1993) Desmoplastic infantile ganglioglioma and desmoplastic cerebral astrocytoma of infancy. Brain Pathol 3:275–281
Conflict of interest
The authors declare that they do not have any conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Ghassan Abuharbid and Majid Esmaeilzadeh contributed equally to this work.
Rights and permissions
About this article

Cite this article
Abuharbid, G., Esmaeilzadeh, M., Hartmann, C. et al. Desmoplastic infantile astrocytoma with multiple intracranial and intraspinal localizations at presentation. Childs Nerv Syst 31, 959–964 (2015). https://doi.org/10.1007/s00381-015-2715-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-015-2715-5