
Abstract
Background
Subependymal giant cell astrocytoma (SEGA) is the most common central nervous system tumor in patients with tuberous sclerosis complex (TSC). SEGAs are generally benign, non-infiltrative lesions, but they can lead to intracranial hypertension, obstructive hydrocephalus, focal neurologic deficits, and even sudden death.
Discussion
Surgical resection has been the standard treatment for SEGAs, and it is generally curative with complete resection. However, not all SEGAs are amenable to safe and complete resection. Gamma Knife stereotactic radiosurgery provides another treatment option as a primary or adjuvant treatment for SEGAs, but it has highly variable response effects with sporadic cases demonstrating its efficacy. Recently, biologically targeted pharmacotherapy with mammalian target of rapamycin (mTOR) inhibitors such as sirolimus and everolimus has provided a safe and efficacious treatment option for patients with SEGAs. However, SEGAs can recur few months after drug discontinuation, indicating that mTOR inhibitors may need to be continued to avoid recurrence. Further studies are needed to evaluate the advantages and adverse effects of long-term treatment with mTOR inhibitors. This review presents an overview of the current knowledge and particularly highlights the surgical and medical options of SEGAs in patients with TSC.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Subependymal giant cell astrocytoma (SEGA) is a tumor typically occurring in the lateral ventricle near the foramen of Monro. SEGA is the most common central nervous system neoplasm in patients with tuberous sclerosis complex (TSC), which is a genetic, multisystem disorder caused by a mutation in the tumor suppressor genes TSC1 and TSC2. Although there have been case reports of solitary SEGAs occurring in the absence of any other TSC-related lesions, these cases are likely due to somatic mosaicism involving the TSC gene [1, 2]. SEGAs commonly occur mostly in the first two decades of life, with average age of presentation being 11 years, but they have also been reported later [3], in the first year of life [4] and even as early as 19 weeks of gestation [5]. The common clinical presentations of patients with SEGAs include seizures, mental retardation, cognitive disability, visual disturbance, headache, and vomiting. Histopathologically, recent reports have demonstrated that SEGAs possess mixed glioneuronal nature at the immunohistochemical and ultrastructure level. SEGAs are generally benign, non-infiltrative lesions classified as World Health Organization grade I [6], yet more aggressive lesions can infiltrate the thalamus, hypothalamus, and basal ganglia, and produce significant perilesional vasogenic edema. Moreover, due to their special location and growth potential, SEGAs can lead to intracranial hypertension, obstructive hydrocephalus, focal neurologic deficits, and even sudden death [7]. Furthermore, massive hemorrhages and malignant changes have also been described in association with SEGAs [8, 9]. Surgical resection has been the standard treatment for SEGAs, and it is generally curative with gross total tumor excision. Although Gamma Knife stereotactic radiosurgery (GKS) has also been used for primary or adjuvant treatment, its specific role in SEGAs still remains unclear. Recently, inhibitors of the mammalian target of rapamycin (mTOR) have been demonstrated to significantly decrease tumor volume and seizure frequency in SEGA patients associated with TSC [10, 11]. Everolimus, an inhibitor of mTOR, has been approved by the FDA as the first pharmacotherapy alternative to surgery for the TSC-associated SEGAs in patients who require therapeutic intervention but are not candidates for curative surgical resection. In this review, we highlight the current standards in diagnosis, histopathology, and clinical management.
Historical overview and epidemiology
The term “Subependymal giant cell astrocytoma” (SEGA) was coined by Russell et al. to differentiate it from other types of intracranial neoplasms, as it had previously received numerous names, such as astrocytoma, ependymoma, spongioblastoma, and possible ganglioglioma [12]. Tuberous sclerosis complex (TSC) is a genetic disorder that can cause growth of benign tumors in multiple organs, and it affects approximately 1 in 6,000 individuals. SEGAs are found in 5–20 % of patients with TSC and constitute over 90 % of intracranial tumors associated with TSC [13–15]. Most patients suffering from this tumor show clinical and pathological symptoms between 8 and 19 years of age [15]. However, there are also reported cases that occurred at the later ages [16] and especially during the early ages of childhood [4]—many of them diagnosed in the prenatal or neonatal age and even as early as 19 weeks of gestation [5]. It can also take place with no relation to family history of TSC, as these would be scattered cases [17]. SEGAs almost exclusively occur in the lateral ventricle near the foramen of Monro and rarely at other sites, such as the fourth ventricle and the third ventricle [18]. SEGAs are responsible for 25 % of the excess mortality attributable to TSC [19, 20]. About 10 % of SEGAs originate in the subependymal nodules, usually bilateral, located near the foramina of Monro [15]. The tumor is frequently located only on one side, but it may also appear on both sides simultaneously, even several years apart.
Radiological patterns
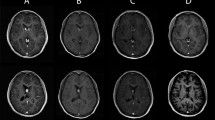
The detection of SEGAs is currently possible using several imaging methods, such as CT or MRI and ultrasound, from the neonatal period or even during gestation [4]. CT or MRI examinations have revealed that almost all SEGAs are located in the vicinity of the foramen of Monro, while some reported that it could also occur in the third ventricles. On CT, the tumors appear as uniform-density masses and high-density calcification in the perimeter of masses. MRI represents the main diagnostic modality for SEGAs. On MRI, the tumors show iso- or hypointensity on T1WI and iso- or hyperintensity on T2WI [4, 16]. Punctuate or nodular calcifications are common in the border of the tumors, while small cystic changes in the tumors are rare [16]. After the injection of contrast agent, the tumors markedly show homogeneous or heterogeneous enhancement. With CT and MRI, together with the specific tumor location, the preliminary diagnosis of SEGAs can be considered (Fig. 1).

On CT, the bilateral tumor shows uniform-density masses and high-density calcification (A and B). On MRI, the tumor shows isointensity on T1WI and T2WI (C and D) and markedly homogeneous enhancement after injection of contrast agent (E)
Histopathology
SEGAs are believed to originate from subependymal nodules of TSC by sequential neuroimaging follow-up [21]. The differentiation between subependymal nodules and SEGA involves size and position rather than histology. Histologically, initial studies that suggested the pure astrocytic nature of SEGAs have been negated by many recent reports which have demonstrated their mixed glioneuronal nature [22, 23]. Indeed, SEGAs are composed of three types of cells: fibrillated spindle cells, swollen gemistocytic-like cells, and giant ganglion-like cells. Appearance of perivascular inflammatory cells that are usually an admixture of mast cells and T lymphocytes is also a common characteristic of SEGAs [24]. Vascularization, angiocentric architectural pattern, and calcification are present with different degrees in some SEGAs [24, 25]. In some SEGAs, mitosis and necrosis can be observed, which do not indicate malignancy in these tumors. Nuclear pseudoinclusions and rosette formations are also present in some cases. At the ultrastructure level, SEGA cells have some features suggestive of neuronal differentiation, including the presence of microtubules, abundant rough endoplastic reticulum cysternae, free ribosomes, and a synapse between a tuber giant cell and an axonal terminal [25]. Indicators of glial differentiation are the presence of bundles of intermediate filaments [25].
Immunohistochemistry
Many recent IHC studies have demonstrated that SEGAs display neuronal and glial cell differentiations. Several neuronal markers, such as neuron-specific enolase (NSE), neurofilament (NF), neuronal cell adhesion molecule (NCAM), substance P (SP), and S-100, and a glial marker, such as glial fibrillary acidic protein (GFAP), are expressed in SEGAs. NSE is expressed in the early stages of the neuronal differentiation process [26]. GFAP is observed in the cells committed to astroglial cells [27]. Anna et al. [25] reported a series of nine SEGAs with immunohistochemistry and showed NF (89 %), NSE (100 %), SP (89 %), and GFAP (100 %) positive. Similarly, Heon et al. [26] also reported a series of eight SEGAs and showed immunoreactivity for NeuN (100 %), NSE (88 %), NCAM (63 %), nestin (100 %), SNP (weakly and focally, 100 %), and GFAP (100 %). Moreover, in an immunohistochemical study with 23 SEGAs reported by Sharma et al. [24], NSE and NF positivity were observed in 15 (65.2 %) cases each. GFAP and S-100 protein positivity, which were more strongly positive in the spindle cells than in other components, were seen in the 23 cases (100 %), varying from focal to diffuse positivity. These immunohistochemical studies combined with ultrastructural studies suggested that the histogenesis of SEGAs might be the stem cells being able to differentiate into both neuronal and glial phenotypes [24–26].
Diagnosis
In the diagnostic process of SEGAs, the most difficult part is to differentiate them from subependymal nodules (SEN), which is also one of the three major brain lesions and the incidence is 92.5–100 % in TSC [28]. Nodules and tumors are both located at the subependymal region. Histopathologically, SEN and SEGA are even described as indistinguishable [14, 29] due to the fact that both lesions are of mixed glioneuronal lineage and contain giant cells. Therefore, the identification of SEGA and SEN particularly relies on radiological criterias. However, till now, radiological features in distinguishing both lesions are also difficult especially at an earlier stage when the lesion is very small [14, 28]. At present, radiological criterias defining SEN and SEGA are highly inconsistent. Previously, the identification standard of SEN and SEGAs was focused on their contrast-enhancing characteristics [14]. Now, many researchers have pointed out that signal intensity and contrast enhancement are not convincing enough, taking into account the fact that some SENs might have slight enhancement on CT and MRI [14]. The diagnosis of SEGAs tends to be coupled with the clinical criteria. If a patient has hydrocephalus or increased intracranial pressure, the perimonro lesion could be diagnosed as a tumor undoubtedly. When a perimonro lesion is not big enough to be diagnosed as a tumor, serial follow-up is essential [14]. With the growth of lesion on serial MRI, the diagnosis could be made easily. Currently, on the basis of the radiological feature, that is, a markedly contrast-enhancing lesion on CT and/or MRI around the foramen of Monro [30], combined with the criteria put forward by Cuccia in 2003 which include [14, 28] the presence of hydrocephalus, lesions with diameter greater than 10 mm, new focal neurological deficit attributable to tumor, and/or symptoms of IICP, the diagnosis of SEGAs could be made with more confidence.
Tumorigenesis
The development of hematomas in TSC fits a two-hit mutational mechanism that was first proposed by Knudson in 1970 for retinoblastoma oncogenesis and subsequently is widely used to explain tumorigenesis in TSC. The first hit corresponds to a germline mutation (spontaneous or inherited) inactivating one allele of either TSC1 or TSC2, and the second hit, called loss of heterozygosity (LOH), is a somatic mutation inactivating the second allele. This two-fit model applies perfectly to most of the hematomas in TSC, but it is relatively rare in SEGAs due to the rare LOH [31]. However, this issue is controversial, as some studies have demonstrated that SEGAs in TSC may be derived from a LOH mechanism, leading to activation of the mTOR kinase [1, 32]. Thus, other possibilities of TSC1/TSC2 complex inactivation have been mooted to explain the pathogenesis of brain lesions including SEGAs in TSC [33–35]. Many reports have demonstrated that post-translational inactivation of the TSC1/TSC2 complex arises through Akt kinase activation [36]. Akt, a downstream effector of PI3K (phosphoinositide-3-OH kinase), is known to activate mTOR-dependent translation. The other mechanism of tumorigenesis of SEGAs in TSC is the inactivation of the hamatrin–tuberin complex after phosphorylation by various kinases, such as extracellular signal-regulated kinase (ERK) [34]. ERK activation has been commonly detected in SEGAs and might be a molecular trigger for their development [34]. In addition, the stability of TSC1/TSC2 complex may also be affected by death-associated protein kinase (DAPK), which functions in a wide range of biological pathways, including TNF-regulated cell death, stress-induced apoptosis, and autophagy [37]. Immunohistochemical studies reveal that mTOR activation appears to be a central event in the pathogenesis of SEGAs; increased levels of different proteins that have been shown to indicate mTOR activation have been detected in cultured TSC1 and TSC2 null cells [38]. Recently, gene expression profiling of SEGAs has indicated that other genes are possibly associated with the development of these tumors. Some genes involved in tumorigenesis and nervous system development, like ANXA1, GPNMB, LTF, RND3, S100A11, SFRP4, and NPTX1, are likely to be mTOR effector genes in SEGAs, as their expression was modulated by an mTOR inhibitor, rapamycin, in SEGA-derived cells [39]. Figure 2 summarizes the role of the mTOR complex and the subsequent downstream cascade that leads to the development of SEGAs and seizures in TSC.

This scheme summarizes the role of mTOR activation in the development of SEGAs and seizures in TSC. IGF-1R insulin-like growth factor-1 receptor, Ras rat sarcoma, Abl Abelson kinase, Akt protein kinase B, PI3 K phosphoinositide 3-kinase, PTEN phosphatase and tensin homologue, TSC1 tuberous sclerosis complex 1, TSC2 tuberous sclerosis complex 2, Rheb Ras homologue enriched in the brain, mTOR mammalian target of rapamycin, S6K1 40 S ribosomal S6 kinase, S6 ribosomal protein S6, eIF4B eukaryotic translation initiation factor 4B, 4EBP1 eukaryotic translation initiation factor 4E binding protein 1, eIF4G eukaryotic translation initiation factor 4G, eIF4E eukaryotic translation initiation factor 4E
Clinical presentation and natural course
Common clinical presentations of patients with SEGAs include seizures (generalized tonic clonic seizures or focal motor seizures), mental retardation, cognitive disability, visual disturbance (decreased vision, diplopia, or blindness), headache, and vomiting. Epilepsy is the most common neurological symptom in TSC with incidence of 60–96 % [40, 41] and often quite severe and unremitting. Mental retardation is common, affecting 40–80 % of TSC patients [42–44]. The distribution of intelligence quotient (IQ) in TSC appears to be biphasic, with one peak around 20 and another peak around 80 [45]. Cognitive disability tends to be moderate or severe in degree [45]. Visual disturbance, headache, and vomiting are symptoms of intracranial hypertension or hydrocephalus, caused by the obstruction of CSF pathway due to SEGAs, which are localized at perimonro region on one or both sides [46]. SEGAs are seen mainly in children and adolescents and associated with male predominance [24].
Serial neuroimaging studies have shown that SEGAs usually correspond to growing SENs, which is supported by the coexistence of pathological specimen intermediate between nodules and SGCA [21]. Transformation of a SEN into a SEGA is usually a gradual process, of which the highest rate is in the first two decades of life [14, 47]. The growth of SEN peaks at puberty and stops by the end of the third decade of life [48]. The minimum interval between detection and significant growth of SENs ranges from 1 to 3 years [19, 47]. Nabbout put forward the risk factors of transformation of a SEN into a SEGA: diameter above 5 mm, incomplete calcification at the perimonro region, and enhancement after gadolinium administration [19].
SEGAs usually continue to grow naturally before intervention with diverse growth rates. Jiang and colleagues demonstrated continuous growth of tumors in the preoperative period in their two patients and calculated the rates as 4.1 and 5.6 mm/year [49]. Park and coworkers also reported four patients with SEGAs and the median growth rate was 2.53 mm/year [50]. Moreover, Cuccia and coworkers [28] reported a mean growth rate in diameter of 3.4 mm/year (range 1–10 mm) in their 15 consecutive patients with SEGAs. They suggested that hydrocephalus is inevitable in patients with SEGAs over time because they observed that no tumor ceased growing during their follow-up.
Although SEGAs are defined as benign CNS neoplasms, classified as WHO Grade I [51], there clinical course is not always benign. Sudden death because of IICP or hydrocephalus is reported in published literature [7, 21]. In addition, massive hemorrhages and malignant changes have also been described in association with these tumors [8, 9]. Hence, according to the guidelines of the NIH Consensus conference, children with a diagnosis of TSC should have a brain MRI performed every 1 to 3 years, generally up to 21 years of age [52, 53]. Once a probable SEGA is detected, brain MRI should be performed more frequently [14].
Surgical management
Currently, early surgical resection still remains the standard treatment for SEGAs demonstrating serial growth on neuroimaging, particularly when there is evidence of progressive ventriculomegaly or symptomatic hydrocephalus. A progressive attitude that a precocious surgical resection may be considered for asymptomatic children with SEGAs before the appearance of increased intracranial pressure signs has been developed. Outcome can be good when resection is early and total. As SEGAs are benign lesions, a complete and safe removal whenever possible means an almost complete cure [46, 54].
The specific timing of surgical treatment for SEGAs still remains controversial. Historically, the indications for surgical resection have centered around evidence of tumor progression in the setting of symptoms of hydrocephalus or ventriculomegaly on surveillance neuroimaging. Although there are reports of asymptomatic SEGAs ranging from 10 to 22 mm in diameter without associated ventriculomegaly, there is no consensus on indications for surgery based on lesion size alone [14, 28, 46]. Even so, a better outcome has been reported for younger patients; in one study, complications occurred in all the patients who were older than 11 years at the time of surgery, whereas the outcome was excellent in all children younger than 11 years [14]. Moreover, several corroborating reports have demonstrated increased surgical morbidity and mortality in patients with symptomatic intracranial hypertension or massive hydrocephalus at the time of surgery [14, 46, 55]. Furthermore, data from retrospective series suggests that early surgery for small asymptomatic lesions identified by surveillance neuroimaging reduces surgical morbidity and recurrence rates [14, 28].
The surgical approach for SEGAs is determined by tumor extension and the presence of an associated hydrocephalus. Traditionally, transcortical, transventricular, and transcallosal interhemispheric approaches are the most commonly used ones [54]. Recently, with significant advancements in endoscopic techniques and instrumentation, minimally invasive endoscopic procedure is recommended for cystic or solid intraventricular lesions, including SEGAs [56]. Endoscopic procedure is considered as a safe, effective, and less invasive approach with lower morbidity [46, 57]. However, endoscopic procedure is limited to lesions with a diameter of 2 cm or less [3, 58, 59] because it results in a conspicuous increase in operative time with larger lesions, which negates one of the key benefits.
The complications after surgical resection of SEGAs resemble those of any tumor surgery within the cerebral ventricles and around the foramen of Monro. The common complications are transient or permanent motor deficits, hemorrhage, or compressive subdural collection, which have been reported in about 10–20 % of the patients who underwent surgery [14, 28, 46, 60]. Acute post-operative fatal hydrocephalus, a rare complication that is generally secondary to infection or hemorrhage, may also occur [46, 60]. In recent reports, the perioperative mortality rate of SEGAs is as low as 0–10.5 % [28, 46]. Since major complications tend to occur more frequently in patients who are symptomatic for increased intracranial pressure or major hydrocephalus before surgery [14, 55], a SEGA should be resected as soon as clear evidence of growth on two subsequent images are determined [46]. For infiltrative or large lesions where gross total resection is nearly impossible to be achieved safely, progression of the residual tumor commonly occurs within follow-up. The authors of the previous surgical series have reported that total resection was achieved in 57–88 % of the cases [28, 46, 49, 55]. In one surgical series of 19 patients with SEGAs, gross total tumor resection was not achieved in four patients and within follow-up, they all received re-operation at a mean of 11.5 months due to tumor progression and/or worsening hydrocephalus [46]. The remaining 15 patients with gross total resection did not experience tumor recurrence with median follow-up of 69 months. For patients with incomplete tumor resection, a close clinical and radiological follow-up is essential for a longer period because many patients experience a recurrence during follow-up, leading to subsequent re-operation [28, 48, 55, 60]. Partial excision of SEGAs could remain steady over years; however, they almost invariably recur, which is sometimes fatal [23, 55, 61].
Gamma Knife stereotactic radiosurgery
GKS has revealed good outcomes for many types of benign brain tumors, including gliomas, with a low incidence of side effects [62], but its role in SEGAs is only reported in scattered cases and therefore so far has not been well affirmed. Among these scattered cases, GKS has been reported as both a primary and adjuvant treatment for SEGAs with highly variable response effects. In a study of GKS treatment for two patients with SEGAs, the tumor volume reduced 70 and 80 %, respectively, within 6 months [63]. However, Wang reported GKS treatment for three patients with SEGAs, and all tumors progressed with median follow-up of 67 months [64]. Subsequently, Henderson reported two patients with SEGAs receiving GKS, and one SEGA was controlled and the other progressed [62]. Most recently, in a study of six patients with SEGAs without hydrocephalus who received GKS, local tumor control was achieved in four patients with progression-free intervals of 24, 42, 57, and 66 months [65]. For the four patients with tumor control, the reductions in tumor volumes were 0, 59, 63, and 86 %, respectively. Of the six patients, four patients received GKS as primary therapy, one of whom progressed and ultimately required surgical excision. In view of slow tumor volume reduction after GKS, this treatment is not recommended for patients with definite evidence of hydrocephalus or progressive ventriculomegaly. Moreover, the application of radiosurgery is further complicated by the fact that radiation of SEGAs may promote malignant degeneration and development of glioblastoma [66, 67]. Even though the role of GKS in SEGAs is limited by the sporadic cases, these reported results suggest GKS may be an additional option for SEGAs that are small but progressively enlarging tumors where complete resection has not been safely achieved, residual or recurrent tumors. Certainly, the precise efficacy and adverse effect of GKS on SEGAs require larger patient populations and longer follow-up.
Pharmacotherapy
In recent years, perhaps the most significant progress in the treatment of SEGAs has derived from the identification of mTOR as the key protein kinase that is inhibited in patients with TSC. TSC is an autosomal dominant disorder caused by the inactivation in one of two tumor suppressor genes, TSC1 or TSC2. Under normal conditions, the TSC1 gene on chromosome 9q34 encodes the protein hamartin, while TSC2, located on chromosome 16p13.3, encodes tuberin. Hamartin–tuberin complex, activating a GTPase, inhibits phosphorylation of the small G protein “Ras-homolog-enriched in brain” (Rheb) that directly activates mTOR [60, 68]. When a TSC1 or TSC2 mutation is present, hamartin–tuberin complex cannot form and mTOR is constitutively over activated, leading to unregulated cell growth, proliferation, metabolism, and angiogenesis [68]. Therefore, constitutive over activation of mTOR becomes an attractive target for pharmacotherapy. The mTOR inhibitors rapamycin (sirolimus) along with the prodrug CCI-779 (temsirolimus) and the analog RAD001 (everolimus) are presently under active investigation for a wide array of oncology indications, including the treatment of TSC-associated SEGAs and as drug-resistant for TSC-associated epilepsy [69].
The first study with rapamycin (sirolimus) in four patients with SEGAs was published in 2006 and demonstrated a significant reduction in SEGA volume, with 46 to 63 % reduction in tumor volume within 2.5 to 5 months at doses commonly used in transplant medicine (trough levels, 10 to 15 ng/mL) [70]. Its efficacy in SEGAs has been subsequently confirmed in later studies [10, 71–73].
More recently, with encouraging preliminary results with rapamycin, a phase 2 open-label clinical trial using everolimus to treat SEGAs in 28 patients with TSC showed SEGA volume reduction of at least 30 % in 21 patients (75 %) and at least 50 % in nine patients (32 %) within the initial 6 months [11]. The therapy with everolimus was continued for a median duration of 21.5 months with trough concentrations of 5 to 15 ng/mL. Importantly, none of the patients treated with mTOR inhibitors required surgical intervention or developed new lesion. Moreover, the treatment was also associated with a reduction in the overall seizure frequency, a relief of CSF obstruction, and an improvement in quality of life. Immediately after the open-label phase 2 study of everolimus, a double-blind, randomized, placebo-controlled phase 3 trial with 117 SEGA patients was carried out, and the result published showed that the trial met its primary endpoint with 35 % of everolimus-treated patients experiencing 50 % or greater reduction in total SEGA volume compared with 0 % reduction in the placebo group (p < 0.0001) [18]. Different degrees of reduction of SEGA size have been observed in all the 155 patients treated with mTOR inhibitors (Table 1). Everolimus has now received US Food and Drug Administration approval for treatment of patients with SEGA associated with TSC that are not amenable to surgical resection.
Common side effects in SEGAs with mTOR inhibitors include upper respiratory infections, stomatitis, aphthous ulcers, acneiform rash, diarrhea, arthralgias, thrombocytopenia, elevations of serum cholesterol, and lipoproteins [11, 69, 70, 73]. These side effects which are often self-limited and generally considered accepted are linked to immunosuppression and mild derangements of lipid metabolism.
Unfortunately, both case reports of sirolimus and the phase 2 trial of everolimus have demonstrated tumor recurrence a few months after drug discontinuation; although the drug can lead to a marked regression of SEGAs [11, 70]. Hence, questions of the optimal drug duration and dosage arise. The duration of drug treatment might be continuous until the patients reach 30 years, if these patients experience spontaneous stabilization of their lesions. For some patients without symptoms until 30 years old, lifelong drug treatment with mTOR inhibitors may be required. Short-term treatment with mTOR inhibitors is quite safe, and short-term side effects are generally accepted; however, long-term side effects are little known when chronically administered for decades or more. Long-term efficacy and safety of low-dosage of mTOR inhibitors should be evaluated with future studies.
Epilepsy
Epilepsy is a hallmark of TSC, present in up to 90 % of patients and commonly refractory to treatment [74]. TSC-associated epilepsy commonly begins during the first year of life and is often focal initially [75]. Infantile spasms, the other major type of early seizures in TSC might precede, coexist, or follow focal seizures during the first months [76]. Early seizure onset, mainly infantile spasms, is related to an increased risk of neurodevelopment and cognitive impairment [75]. TSC2 mutation tends to be related with earlier seizure onset than TSC1 mutation. Seizures tend to start at an older age in familial TSC than in non-familial cases [77].
Despite treatment with multiple antiepileptic drugs (AEDs), there is often an increase in seizure severity and frequency resulting in significant developmental delay and cognitive impairment, which may necessitate surgical intervention for seizure control. Currently, most AEDs primarily function by decreasing neuronal activity via direct modulation of ion channels or neurotransmitter receptors, such as sodium channels and GABAA receptors [78]. From this, mTOR inhibitors do not appear to act like standard AEDs. Nevertheless, while it is unlikely that mTOR inhibitors bind directly to ion channels, the mTOR pathway could be related to regulating the expression of ion channels via effects on protein translation, which might subsequently reduce neuronal excitability. For example, mTOR inhibitors have been indicated to increase the expression of potassium channels and decrease the expression of glutamate receptors [79, 80]. Moreover, both case reports and clinical trials have demonstrated a significant reduction in seizure frequency and AED requirements in TSC patients treated with mTOR inhibitors [68–70]. In some cases, the response has been profound with patients remaining seizure free for months after tapering and/or discontinuation of AEDs. This reduced seizure burden may lead to improved cognitive performance and quality of life. Preliminary results from this uncontrolled trial also indicate that mTOR inhibitors can decrease seizure frequency in a subset of TSC patients; 9 out of 17 (53 %) patients had a greater than 50 % reduction in seizures, including three patients that became seizure free [81]. In phase 2 study including 28 patients with SEGA, everolimus treatment decreased seizure frequency in 56 % of the 16 patients evaluated with 24-h video EEG between inclusion and 6 months [11]. The above observations in humans are further supported by animal models of TSC, where rapamycin treatment can prevent the development of seizures and reverse learning defects [82]. Improved seizure control and the ability to minimize or discontinue AEDs with mTOR treatment are significant. While these preliminary data are encouraging, long-term studies are necessary to investigate seizure control, safety, and neuronal development with mTOR inhibitors.
Lesson learned and future directions
Early surgical resection has been, and is presently, the standard treatment for SEGAs demonstrating serial growth on neuroimaging, particularly when there is evidence of progressive ventriculomegaly or symptomatic hydrocephalus. A progressive attitude that a precocious surgical resection may be considered for asymptomatic children with SEGAs before the appearance of increased intracranial pressure signs is being adopted to decrease the morbidity and mortality rates. However, the dramatic response of TSC-associated SEGAs to mTOR inhibitors suggests that these drugs could be a potential alternative to surgery.
GKS may be an additional option for SEGAs that are small but progressively enlarging tumors where complete resection has not been safely achieved and in residual or recurrent tumors. However, the precise efficacy and adverse effect of GKS on SEGAs require larger patient populations and longer follow-up.
Currently, the therapies for SEGAs are mainly focused on surgery and medication. Both the open-label clinical trial and case reports have demonstrated that mTOR inhibitors, such as sirolimus and everolimus, can obviously decrease SEGA volume and improve ventriculomegally and symptoms of obstructive hydrocephalus, which is a promise for an effective medical treatment of SEGA [11, 71, 73, 80]. Currently, everolimus is approved in the USA for patients with SEGAs that are not amenable to surgical resection. Medical treatment with mTOR inhibitors could be recommended for asymptomatic SEGAs with the radiographic evidence of tumor progression. mTOR inhibitors could also be used to facilitate sequent surgery for a SEGA that presents invasion into deep white matter structures, bilateral lesions, or in an atypical location. Furthermore, in case of re-growth after the first resection, mTOR inhibitors could be used to keep lesion size under control in consideration of the higher risk of a second surgery. However, both case reports of sirolimus and the phase 2 trial of everolimus have demonstrated tumor recurrence a few months after drug discontinuation. Hence, questions of the optimal drug duration and dosage arise, waiting for further studies. The duration of drug treatment might be chronic or lifelong; long-term efficacy and safety of mTOR inhibitors in TSC-associated SEGAs also need to be evaluated. When choosing between surgical and/or medical treatment, the risks and benefits of each option should be considered thoroughly.
Recently, gene expression profilings show that some genes involved in tumorigenesis and nervous system, like ANXA1, GPNMB, LTF, RND3, S100A11, SFRP4, and NPTX1, are all modulated by mTOR inhibitors [39]. However, in addition to mTOR, ERK signaling pathway is also activated in TSC associated with SEGAs [45, 83, 84]. mTOR hyperactivity is responsible for cell overgrowth [85]; rapamycin does not influence either SEGA cell migration or actin cytoskeleton arrangement, but it decreases SEGA cell size. However, ERK signaling has been shown to control remodeling of acting cytoskeleton and cell migration [86, 87], which is further supported by the observation that inhibition of ERK signaling with U0126 significantly diminishes proliferation and cell migration of SEGA cells but does not influence SEGA cell size. Combined suppressions of mTOR and ERK signaling pathways brought forth the most remarkable decrease of SEGA cell viability, proliferation, and actin cytoskeleton rearrangement. These results suggest that mTOR and ERK signaling pathways may be responsible for pathogenesis and progression of SEGA. Medical treatment targeted at both mTOR and ERK signaling pathways may give better results in the treatment of SEGAs than using mTOR inhibitors alone.
Abbreviations
- SEGA:
-
subependymal giant cell astrocytoma
- TSC:
-
tuberous sclerosis complex
- GKS:
-
Gamma Knife stereotactic radiosurgery
- mTOR:
-
mammalian target of rapamycin
- SEN:
-
subependymal nodules
- LOH:
-
loss of heterozygosity
- ERK:
-
extracellular signal-regulated kinase
- DAPK:
-
death-associated protein kinase
- AEDs:
-
antiepileptic drugs
Reference
Ichikawa T, Wakisaka A, Daido S, Takao S, Tamiya T, Date I (2005) A case of solitary subependymal giant cell astrocytoma: two somatic hits of TSC2 in the tumor, without evidence of somatic mosaicism. J Mol Diagn 7:544–549
Kwiatkowska J, Wigowska-Sowinska J, Napierala D, Slomski R, Kwiatkowski DJ (1999) Mosaicism in tuberous sclerosis as a potential cause of the failure of molecular diagnosis. N Engl J Med 340:703–707
Souweidane MM, Luther N (2006) Endoscopic resection of solid intraventricular brain tumors. J Neurosurg 105:271–278
Hahn JS, Bejar R, Gladson CL (1991) Neonatal subependymal giant cell astrocytoma associated with tuberous sclerosis: MRI, CT and ultrasound correlation. Neurology 41:124–128
Medhkour A, Traul D, Hussain N (2002) Neonatal subependymal giant cell astrocytoma. Pediatr Neurosurg 36:271–274
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropsychiatrica 114:97–109
Byard RW, Blumbergs PC, James RA (2003) Mechanisms of unexpected death in tuberous sclerosis. J Forensic Sci 48:172–176
Brown JM (1975) Tuberose sclerosis with malignant astrocytoma. Med J Aust 1:811–814
Waga S, Yamamoto Y, Kojima T, Sakakura M (1977) Massive hemorrhagein tumor of tuberous sclerosis. Surg Neurol 8:99–101
Koenig MK, Butler IJ, Northrup H (2008) Regression of subependymal giant cell astrocytoma with rapamycin in tuberous sclerosis complex. J Child Neurol 23:1238–1239
Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T, Franz DM (2010) Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med 363:1801–1811
Russell DS, Rubenstein LJ, Lumsden CE (1959) Tuberose sclerose (Bourneville’s disease), subependymal giant-cell astrocytomas: tuberose sclerosis, spongio neuroblastoma and tuberose sclerosis. In: Russell DS, Rubenstein LJ, editors. Pathology of tumours of the nervous system. London: Edward Arnold p. 29-30. 105–6, 169
Adriaensen ME, Schaefer-Prokop CM, Stijnen T, Duyndam DA, Zonnenberg BA, Prokop M (2009) Prevalence of subependymal giant cell tumors in patients with tuberous sclerosis and a review of the literature. Eur J Neurol 16:691–696
Goh S, Butler W, Thiele EA (2004) Subependymal giant cell tumors in tuberous sclerosis complex. Neurology 63:1457–1461
Shepherd CW, Gómez MR, Lie JT, Crowson CS (1991) Causes of death in patients with tuberous sclerosis. Mayo Clin Proc 66:792–796
Takata K, Gaspareto EL, Da Costa LC, Lucato LT, Reed UC, Matushita H, de Aguiar PHP, Rosemberg S (2007) Subependymal giant cell astrocytoma in patients with tuberous sclerosis: magnetic resonance imaging findings in ten cases. Arq Neuropsiquiatr 65:313–316
Raju GP, Urion DK, Sahin M (2007) Neonatal subependymal giant cell astrocytoma: new case and review of literature. Pediatr Neurol 36:128–131
Franz DN, Belousova E, Sparagana S, Bebin EM, Frost M, Kuperman R (2012) Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet, Available online 13 November
Nabbout R, Santos M, Rolland Y, Delalande O, Dulac O, Chiron C (1999) Early diagnosis of subependymal giant cell astrocytoma in children with tuberous sclerosis. J Neurol Neurosurg Psychiatry 66:370–375
Sinson G, Sutton LN, Yachnis AT, Duhaime AC, Schut L (1994) Subependymal giant cell astrocytomas in children. Pediatr Neurosurg 20:233–239
Fujiwara S, Takaki T, Hikita T, Nishio S (1989) Subependymal giant-cell astrocytoma associated with tuberous sclerosis. Do subependymal nodules grow? Childs Nerv Syst 5:43–44
Debiec-Rychter M, Jesionek-Kupnicka D, Zakrzewski K, Liberski PP (1999) Cytogenetic changes in two cases of subependymal giant-cell astrocytoma. Cancer Gente Cytogenet 109:29–33
Sharma MC, Ralte AM, Gaekwad S, Santosh V, Shankar S, Sarkar C (2004) Subependimal giant cell astrocytoma—a clinicopathological study of 23 cases with special emphasis on histogenesis. Pathol Oncol Res 10:219–224
Sharma MC, Ralte AM, Arora R, Santosh V, Shankar SK, Sarkar C (2004) Subependymal giant cell astrocytoma—a clinicopathological study of 23 cases with special emphasis on histogenesis. Pathol Oncol Res 10(4):219–224
Anna MB, Alessandro F, Francesca C, Chiara FG, Federico M, Flavio G, Genitori L, Taddei GL (2009) Subependymal giant cell astrocytoma (SEGA): is it an astrocytoma? Morphological, immunohistochemical and ultrastructural study. Neuropathology 29:25–30
Heon Y, Young Im K, Soo Young I, Haeyoung SK, Sun Ha P, Sung-Hye P, Kim DG, Jung HW (2005) Immunohistochemical study of central neurocytoma, subependymoma, and subependymal giant cell astrocytoma. J Neuro-Oncol 74:1–8
Bonnin JM, Rubinstein LJ (1984) Immunohistochemistry of central nervous system tumors. Its contribution to neurosurgical diagnosis. J Neurosurg 60:1121–1133
Cuccia V, Zuccaro G, Sosa F, Monges J, Lubienieky F, Taratuto AL (2003) Subependymal giant cell astrocytoma in children with tuberous sclerosis. Childs Nerv Syst 19:232–243
Shepherd CW, Scheithauer BW, Gomez MR, Altermatt HJ, Katzmann JA (1991) Subependymal giant cell astrocytoma: a clinical, pathological, and flow cytometric study. Neurosurgery 28:864–868
Menor F, Marti-Bonmati L, Mulas F, Poyatos C, Cortina H (1992) Neuroimaging in tuberous sclerosis: a clinicoradiological evaluation in pediatric patients. Pediatr Radiol 22:485–489
Sepp T, Yates JR, Green AJ (1996) Loss of heterozygosity in tuberous sclerosis hamartomas. J Med Genet 33:962–964
Niida Y, Stemmer-Rachamimov AO, Logrip M, Tapon D, Perez R, Kwiatkowski DJ, Sims K, MacCollin M, Louis DN, Ramesh V (2001) Survey of somatic mutations in tuberous sclerosis complex (TSC) hamartomas suggests different genetic mechanisms for pathogenesis of TSC lesions. Am J Hum Genet 69:493–503
Baskin HJ Jr (2008) The pathogenesis and imaging of the tuberous sclerosis complex. Pediatr Radiol 38:936–952
Jozwiak J, Jozwiak S, Wlodarski P (2008) Possible mechanisms of disease development in tuberous sclerosis. Lancet Oncol 9:73–79
Kwiatkowski DJ, Manning BD (2005) Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum Mol Genet 14:251–258
Zhang H, Cicchetti G, Onda H (2003) Loss of Tsc1/Tsc2 activates mTOR and disrupts P13K–Akt signaling through downregulation of PDGFR. J Clin Invest 112:1223–1233
Lin Y, Henderson P, Pettersson S, Satsangi J, Hupp T, Stevens C (2011) Tuberous sclerosis-2(TSC2) regulates the stability of death-associated protein kinase-1(DAPK) through a lysosome-dependent degradation pathway. FEBS J 278(2):354–370
Chan JA, Zhang H, Roberts PS, Jozwiak S, Wieslawa G, Lewin Kowalik J, Katarzyna K, David JK (2004) Pathogenesis of tuberous sclerosis subependymal giant cell astrocytomas: biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. J Neuropathol Exp Neurol 63:1236–1242
Tyburczy ME, Kotulska K, Pokarowski P, Mieczkowski J, Kucharska J, Grajkowska W, Roszkowski M, Jozwiak S, Kaminska B (2010) Novel proteins regulated by mTOR in subependymal giant cell astrocytomas of patients with tuberous sclerosis complex and new therapeutic implications. Am J Pathol 176(4):1878–1890
Cross JH (2005) Neurocutaneous syndromes and epilepsy-issues in diagnosis and management. Epilepsia 46(Suppl 10):17–23
Gomez MR (1999) Natural history of cerebral tuberous sclerosis. In: Gomez MR, Sampson JR, Whittemore VH (eds) Tuberous Sclerosis Complex: Developmental Perspectives in Psychiatry. Oxford University Press, New York, pp 29–46
Jambaque I, Cusmai R, Curatolo P, Cortesi F, Perrot C, Dulac O (1991) Neuropsychological aspects of tuberous sclerosis in relation to epilepsy and MRI findings. Dev Med Child Neurol 33:698–705
O’Callaghan FJ, Harris T, Joinson C, Bolton P, Noakes M, Presdee D, Renowden S, Shiell A, Martyn CN, Osborne JP (2004) The relation of infantile spasms, tubers, and intelligence in tuberous sclerosis complex. Arch Dis Child 89:530–533
Osborne J, Webb D (1993) Seizures and intellectual disability associated with tuberous sclerosis. Dev Med Child Neurol 35:276
Gregory LH, Carl ES, The Tuberous Sclerosis Study Group (2007) Tuberous sclerosis complex and epilepsy: recent developments and future challenges. Epilepsia 48(4):617–630
de Ribaupierre S, Dorfmüller G, Bulteau C, Fohlen M, Pinard JM, Chiron C, Catherine C, Olivier D (2007) Subependymal giant-cell astrocytomas in pediatric tuberous sclerosis disease: when should we operate? Neurosurgery 60:83–89
Moavero R, Pinci M, Bombardieri R, Curatolo P (2011) The management of subependymal giant cell tumors in tuberous sclerosis: a clinician's perspective. Childs Nerv Syst 27:1203–1210
Torres VE, King BF, McKusick MA, Bjornsson J, Zincke H (2001) Update on tuberous sclerosis complex. Contrib Nephrol 136:33–49
Jiang T, Jia G, Ma Z, Luo S, Zhang Y (2011) The diagnosis and treatment of subependymal giant cell astrocytoma combined with tuberous sclerosis. Childs Nerv Syst 27:55–62
Park KJ, Kano H, Kondziolka D, Niranjan A, Flickinger JC, Lunsford LD (2011) Gamma Knife surgery for subependymal giant cell astrocytomas. J Neurosurg 114:808–813
Lopes MBS, Wiestler OD, Stemmer-Rachamimov AO, Sharma MC (2007) Tuberous sclerosis complex and subependymal giant cell astrocytoma, in Louis DN, Ohgaki H, Wiestler OD, Cavenee WK (eds): WHO Classification of Tumours of the Central Nervous System, ed 4. Lyon: IARC Press Vol 1, pp 218–221
Hyman MH, Whittemore VH (2000) National Institutes of Health consensus conference: tuberous sclerosis complex. Arch Neurol 57:662–665
Roach ES, DiMario FJ, Kandt RS, Northrup H (1999) Tuberous Sclerosis Consensus Conference: recommendations for diagnostic evaluation. National Tuberous Sclerosis Association. J Child Neurol 14:401–407
Berhouma M (2010) Management of subependymal giant cell tumors in tuberous sclerosis complex: the neurosurgeon’s perspective. World J Pediatr 6:103–110
Kim SK, Wang KC, Cho BK, Jung HW, Lee YJ, Chung YS, Lee JY, Park SH, Kim YM, Choe G, Chi JG (2001) Biological behavior and tumorigenesis of subependymal giant cell astrocytomas. J Neurooncol 52:217–225
Boogaarts HD, Decq P, Grotenhuis JA, Le GC, Nseir R, Jarraya B, Djindjian M, Michel D, Tjemme B (2011) Long-term results of the neuroendoscopic management of colloid cysts of the third ventricle: a series of 90 cases. Neurosurgery 68:179–187
Cai R, Di X (2010) Combined intra- and extra-endoscopic techniques for aggressive resection of subependymal giant cell astrocytomas. World Neurosurg 73:713–718
Beems T, Grotenhuis JA (2001) Subependymal giant cell astrocytoma in tuberous sclerosis: endoscopic images and the implications for therapy. Minim Invasive Neurosurg 44:58–60
Cappabianca P, Cinalli G, Gangemi M, Brunori A, Cavallo L, deDivitiis E, Decq P, Delitala A, Di Rocco F, Frazee J, Godano U, Grotenhuis A, Longatti P (2008) Application of neuroendoscopy to intraventricular lesions. Neurosurgery 62(suppl2):575–597
Shepherd CW, Gomez MR (1991) Mortality in the Mayo Clinic Tuberous Sclerosis Complex Study. Ann NY Acad Sci 615:375–377
Franz DN, de Vries PJ, Crino PB (2009) Giant cell astrocytomas in tuberous sclerosis complex. Arch Dis Child 94:75–76
Henderson MA, Fakiris AJ, Timmerman RD, Worth RM, Lo SS, Witt TC (2009) Gamma Knife stereotactic radiosurgery for low-grade astrocytomas. Stereotact Funct Neurosurg 87:161–167
Park YG, Kim EY, Chang JW, Chung SS (1997) Volume changes following gamma knife radiosurgery of intracranial tumors. Surg Neurol 48:488–493
Wang LW, Shiau CY, Chung WY, Wu HM, Guo WY, Liu KD, Ho DMT, Wong TT, Pan DHC (2006) Gamma Knife surgery for low-grade astrocytomas: evaluation of long-term outcome based on a 10-year experience. J Neurosurg 105(suppl):127–132
Park KJ, Kano H, Kondziolka D, Niranjan A, Flickinger JC, Lunsford LD (2011) Gamma Knife surgery for subependymal giant cell astrocytomas. Clinical article. J Neurosurg 114:808–813
Matsumura H, Takimoto H, Shimada N, Hirata M, Ohnishi T, Hayakawa T (1998) Glioblastoma following radiotherapy in a patient with tuberous sclerosis. Neurol Med Chir (Tokyo) 38:287–291
Torres OA, Roach ES, Delgado MR, Sparagana SP, Sheffield E, Swift D, Bruce D (1998) Early diagnosis of subependymal giant cell astrocytoma in patients with tuberous sclerosis. J Child Neurol 13:173–177
Perek-Polnik M, Jozwiak S, Jurkiewicz E, Perek D, Kotulska K (2012) Effective everolimus treatment of inoperable, life-threatening subependymal giant cell astrocytoma and intractable epilepsy in a patient with tuberous sclerosis complex. Eur J Paediatr Neurol 16:83–85
Franz DN (2011) Everolimus: an mTOR inhibitor for the treatment of tuberous sclerosis. Expert Rev Anticancer Ther 11:1181–1192
Franz DN, Leonard J, Tudor C, Chuck G, Care M, Sethuraman G et al (2006) Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol 59:490–498
Birca A, Mercier C, Major P (2010) Rapamycin as an alternative to surgical treatment of subependymal giant cell astrocytomas in a patient with tuberous sclerosis complex. J Neurosurg Pediatr 6:381–384
Bissler JJ, McCormack FX, Young LR, Elwing JM, Chuck G, Leonard JM, Schmithorst VJ, Laor T, Brody AS, Bean J, Salisburyand S, Franz DN (2008) Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 358:140–151
Lam C, Bouffet E, Tabori U, Mabbott D, Taylor M, Bartels U (2010) Rapamycin (sirolimus) in tuberous sclerosis associated pediatric central nervous system tumors. Pediatr Blood Cancer 54:476–479
Holmes GL, Stafstrom CE (2007) Tuberous sclerosis complex and epilepsy: recent developments and future challenges. Epilepsi 48:617–630
Chu-Shore CJ, Major P, Camposano S, Muzykewicz D, Thiele EA (2010) The natural history of epilepsyin tuberous sclerosis complex. Epilepsia 51:1236e41
Curatolo P, Bombardieri R, Jozwiak S (2008) Tuberous sclerosis. Lancet 372(9639):657e68
Jansen FE, Braams O, Vincken KL, Algra A, Anbeek P, Jennekens-Schinkel A, Halley D, Zonnenberg BA, Ouweland A, Huffelen AC, Nieuwenhuizen O, Nellist M (2008) Overlapping neurologic and cognitive phenotypes in patients with TSC1 or TSC2 mutations. Neurology 70:908e15
Wong M (2012) mTOR as a potential treatment target for epilepsy. Future Neurol 7(5):537–545
Graham KF, Haddick PC, Jan YN, Jan LY (2006) Activity- and mTOR-dependent suppression of Kv1.1 channel mRNA translation in dendrites. Science 314:144–148
Wang Y, Barbaro MF, Baraban SC (2006) A role for the mTOR pathway in surface expression of AMPA receptors. Neurosci Lett 401:35–39
Wilfong A, Krueger DA, Holland-Bouley K (2011) Everolimus improves seizure control in tuberous sclerosis complex. Presented at: American Epilepsy Society Annual Meeting Baltimore, MD, USA, (Late-breaking Abstract 3.329)
Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ et al (2008) Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med 14:843–848
Govindarajan B, Mizesko MC, Miller MS, Onda H, Nunnelley M, Casper K, Brat D, Cohen C, Arbiser JL (2003) Tuberous sclerosis-associated neoplasms express activated p42/44 mitogen-activated protein (MAP) kinase, and inhibition of MAP kinase signaling results in vivo tumor growth. Clin Cancer Res 9:3469–3475
Jozwiak J, Grajkowska W, Kotulska K, Jozwiak S, Zalewski W, Zajaczkowska A, Roszkowski M, Slupianek A, Wlodarski P (2007) Brain tumor formation in tuberous sclerosis depends on Erk activation. Neuromol Med 9:117–127
Fingar DC, Blenis J (2004) Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 23:3151–3171
Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA (1997) Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol 137:481–492
Pullikuth AK, Catling AD (2007) Scaffold mediated regulation of MAPK signaling and cytoskeletal dynamics: a perspective. Cell Signal 19:1621–1632
Yalon M, Ben-Sira L, Constantini S, Toren A (2011) Regression of subependymal giant cell astrocytomas with RAD001 (everolimus) in tuberous sclerosis complex. Childs Nerv Syst 27:179–181
Acknowledgments
Thanks are given for the important recommendations made by Jian Chen’s colleagues on this article.
Conflict of interest
We certify that there is no actual or potential conflict of interest in relation to this article.
Author information
Authors and Affiliations
Corresponding author
Additional information
Taohui Ouyang and Na Zhang contributed equally to this work.
This article has been retracted by the Journal Editors due to considerable overlap with the article “Advances in the management of subependymal giant cell astrocytoma” by Thomas L. Beaumont, David D. Limbrick, Matthew D. Smyth published in the same journal, Volume 28, no. 7, 2012 pp 963-968.
About this article
Cite this article
Ouyang, T., Zhang, N., Benjamin, T. et al. RETRACTED ARTICLE: Subependymal giant cell astrocytoma: current concepts, management, and future directions. Childs Nerv Syst 30, 561–570 (2014). https://doi.org/10.1007/s00381-014-2383-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-014-2383-x