
Abstract
Tuberous sclerosis complex (TSC) is an autosomal-dominant disorder characterized by the formation of hamartomatous lesions in multiple organ systems. It is the second most common neurocutaneous syndrome after neurofibromatosis type 1 and has been recognized since the late 1800s. Although the disease has complete penetrance, there is also high phenotypic variability: some patients have obvious signs at birth, while others remain undiagnosed for many years. In addition to skin lesions, TSC patients develop numerous brain lesions, angiomyolipoma (AMLs), lymphangiomyomatosis (LAM) in the lungs, cardiac rhabdomyomas, skeletal lesions, and vascular anomalies, all of which are well seen with medical imaging. Our knowledge of TSC genetics and pathophysiology has expanded dramatically in recent years: two genetic loci were discovered in the 1990s and recent elucidation of TSC’s interaction with the mTOR pathway has changed how we manage the disease. Meanwhile, medical imaging is playing an increasingly important role in the diagnosis, management, and treatment of TSC. We provide an update on the genetics and pathophysiology of TSC, review its clinical manifestations, and explore the breadth of imaging features in each organ system, from prenatal detection of cardiac rhabdomyomas to monitoring rapamycin therapy to treatment of AMLs by interventional radiology.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Tuberous sclerosis complex (TSC) is the second most common phakomatosis after neurofibromatosis type 1 [1] and is characterized by the formation of histologically benign hamartomas and low-grade neoplasms in multiple organ systems. It has a prevalence of about 1 in 6,000 newborns and affects approximately 1.5 million people worldwide, occurring in all races and both genders equally. TSC is an autosomal-dominant disorder with high penetrance and variance [2]; obvious signs might be present at birth, while in other patients it eludes diagnosis for decades. Almost all patients with TSC have numerous cutaneous stigmata [3, 4], some of which can be subtle. The vast majority of patients have brain lesions [3] and, although these can cause seizures, learning difficulties, behavioral problems, and autism, only 50% of TSC patients have below average intelligence [5]. Most patients have renal lesions [6], which cause complications in about 10%. Cardiac tumors are usually clinically silent, although they occasionally cause obstruction and arrhythmias [7]. Fewer TSC patients have clinically evident lung disease, and still fewer have vascular complications. Skeletal lesions go largely unnoticed. All of these lesions are very well visualized by medical imaging.
The broad spectrum of pathology seen in TSC is reflected in the expansive history of its elucidation. The disease was first reported in 1862 by Friedrich von Recklinghausen. He described an infant with cardiac rhabdomyomas and sclerotic brain lesions who died shortly after birth, but he did not appreciate how the lesions were related [8]. In 1880, Désiré Bourneville documented the neurologic symptoms of an infant with hard mass lesions in the cortex of the brain that he likened to hard potatoes, coining the term “tuberous sclerosis.” The association of these neurologic abnormalities with skin lesions was made a decade later by John Pringle who described adenoma sebaceum in a patient with mental retardation [9]. During the next century, it became better understood that TSC encompassed abnormalities of the skin, central nervous system (CNS), heart, lungs, kidneys, bones, and blood vessels [10], and the full spectrum of the disease was summarized by Gomez et al. [8, 11] 25 years ago. In the last decade there have been many advances in the molecular genetics and understanding of the pathophysiology of TSC, including the mechanism of action of the TSC1 and TSC2 gene products and benign metastasis of smooth muscle cells causing lymphangiomyomatosis (LAM) [7].
Genetics
TSC is an autosomal-dominant disorder caused by a mutation in either the TSC1 or TSC2 gene [12, 13]. These genes act as tumor suppressors, and a mutation to each allele at one of these loci is required for tumorigenesis, analogous to Knudson’s hypothesis of retinoblastoma oncogenesis [14]. For a patient to express TSC, two mutational events occur: the first is a germ line mutation (spontaneous or familial), resulting in a loss of heterozygosity (LOH) at the TSC1 or TSC2 allele [15–18]; subsequent somatic mutations of the remaining allele result in the phenotype of TSC. Somatic mosaicism [19] can result in a less severe phenotype or a so-called forme fruste variation of the disease [20]. Similarly, it has been discovered that some cases of supposed sporadic TSC1 and TSC2 mutations actually represent inherited defects in nonaffected parents with gonadal mosaicism [20, 21]. TSC1 and TSC2 are found on chromosome 9p and 16q, respectively [12, 13]. TSC1 encodes for a protein called hamartin, which is smaller and thought to help stabilize the gene product of TSC2, tuberin. Hamartin and tuberin form a heterodimer that functions as a tumor suppressor by inhibiting the mTOR kinase cascade. Modulating mTOR activity is important in regulating cell growth and proliferation. Dysfunctional gene products from altered TSC1 or TSC2 gene loci lead to increased activation of mTOR kinase, which then results in disorganized cellular overgrowth and abnormal differentiation [15, 22]. Perhaps because it has a more important role in mTOR regulation, mutations of TSC2 lead to a more severe phenotype [23–25]. TSC1 mutates spontaneously less often than TSC2 and is thus more common in familial cases, but, overall, 70% of TSC mutations are spontaneous, with TSC2 mutations more than three times as common as TSC1 mutations [25].
Although LOH at the TSC loci leads to upregulation of mTOR and subsequent disorganized cell growth, other proposed mechanisms of tumorigenesis do not involve LOH at TSC1 or TSC2, but instead are predicated on the dissociation of the TSC heterodimer, leading to its inactivation. Dissociation of the TSC heterodimer occurs after phosphorylation by various kinases [26]. One important kinase that inactivates the TSC complex is extracellular signal-regulated kinase (ERK) [27], which has been shown to be integral in the formation of subependymal giant cell astrocytomas (SEGAs) and some cardiac rhabdomyomas [28, 29].
Clinical manifestations
The clinical picture traditionally associated with TSC is Vogt’s triad: epilepsy, mental retardation, and facial angiofibromas. This triad is actually seen in only 30–40% of TSC patients; about half of patients have normal intelligence [7]. The presentation of TSC is so variable that some patients are only diagnosed by genetic testing or careful screening after a child or sibling presents with more severe symptoms. For the purposes of this review, the clinical manifestations of TSC are discussed as they relate to imaging findings in the sections that follow, organized by organ system, but a brief discussion of the cutaneous manifestations and diagnostic criteria of TSC is merited.
Cutaneous manifestations
There are many cutaneous stigmata of TSC and they are seen in almost all patients with the disorder [30–32]. The most common and earliest skin finding in TSC is multiple hypopigmented macules (also called ashleaf spots) [31, 32], which are seen in more than 90% of patients before 2 years of age [31]. Hypopigmented macules are not specific for TSC, but a patient with large numbers of such lesions is more likely to have TSC; a Wood’s lamp might be needed to see hypopigmented macules. Several other stigmata are said to be near pathognomonic for TSC, including adenoma sebaceum. This name was first coined by Pringle and is actually a misnomer because the lesion has no sebaceous glands. The lesion is a hamartoma composed of connective and vascular elements and is properly termed an angiofibroma [33]. Angiofibromas form discrete pink papules on the malar region of the face in 70% of TSC patients. Closely related lesions are also found on the forehead of most TSC patients and are termed “forehead plaques.” Shagreen patches are thick, rubbery plaques with a pebbly texture composed of raised connective tissue nevi [32, 34], and are found on the nape of the neck and lumbosacral trunk of 50% of TSC patients. Many older children and adults with TSC have fleshy, raised fibromas around or under the nail bed termed periungual fibromas [34]. Although not cutaneous lesions, dental pits, gingival fibromas, and retinal white patches are also commonly seen in TSC [7]. Less common and less specific lesions also include café-au-lait spots, molluscum fibrosum pendulum, and skin tags.
Diagnosis
The diagnosis of TSC can be difficult because the manifestations are so varied and few signs are specific. For that reason, a set of criteria has been proposed to help clinicians reach a diagnosis. The criteria were revised in 1998 and distinguish between major and minor features. Patients are evaluated and said to have definite, probable, or suspected TSC based on the presence of these criteria (Table 1). Medical imaging is heavily relied upon for most of the major and several of the minor criteria. Genetic testing is also available.
Neurologic manifestations and imaging findings
Neurologic manifestations of TSC are the most common cause of morbidity and mortality in TSC patients. They include seizures, developmental delay, behavioral problems, and autism [35, 36]. Seizures are the most common symptom and occur in 90% of TSC patients. About 50% have mental retardation and these children are more likely to have a TSC2 mutation [5, 7]. Mild to moderate autism is also very common in TSC and present in 25–50% of patients [36]. TSC-associated autism is seen with equal predominance in males and females, as opposed to the male predisposition of autism observed in the general population. It is unclear whether autism is a direct or indirect result of TSC; it might be secondary to temporal lobe tubers or epileptiform foci [36]. Brain imaging findings of TSC include cortical and subcortical tubers, white matter abnormalities, subependymal nodules, and SEGAs.
Cortical and subcortical tubers
Tubers are variably epileptogenic hamartomas that exhibit disorganized cortical lamination. They are formed during embryogenesis and are seen in 90% of TSC patients. The masses can be cortical or subcortical and expand the overlying gyri (Fig. 1). Their gross appearance resembles potatoes, which, with their hard texture, lends the name tuberous sclerosis to this disorder. Pathologically, the hamartomas contain glial and neuronal elements such as atypical giant astrocytes, maloriented neurons, and bizarre giant cells [15]. On CT imaging, tubers are low in attenuation and usually do not enhance [37, 38]. About half calcify [37], which is well seen on CT images and sometimes even on skull radiographs. On MRI, the lesions have low T1 signal and high T2 or FLAIR signal, unless they are calcified, in which case there can be significant signal loss and blooming artefact [37, 38]. The MRI pattern is reversed in young infants, in whom the surrounding unmyelinated brain has increased water content [39]. The imaging characteristics of tubers are also altered when they develop cyst-like changes. Cystic tubers have low T1 and high T2 signal centrally. FLAIR sequences typically show central signal loss with a high signal peripheral rim. The cause of these cystic changes is unknown but might reflect cellular degeneration. It is more commonly seen in children younger than 7 [40]. Proton spectroscopy of tubers generally demonstrates decreased N-acetyl aspartate (NAA) and increased myo-inositol. The former is thought to reflect a reduction of neurons, and increased myo-inositol might be caused by gliosis or immature neurons [41, 42].

Child with cortical and subcortical tubers. Unenhanced CT image (a) and axial FLAIR MR image (b) demonstrate a large subcortical tuber in the left frontal lobe (large arrow) merging with underlying white matter abnormality. Multiple smaller tubers (small black arrows) have a similar CT appearance. These smaller hamartomas, as well as another in the left posterior whiter matter (b white arrow) have a cystic appearance with low-signal intensity centrally and a rim of increased FLAIR signal peripherally. Another child has diffuse mineralization of a left frontal hamartoma that appears as poorly defined, hazy increased attenuation on the CT image (c) and shows corresponding signal loss on the T2-W image (d)
Recently, advanced imaging techniques such as diffusion tensor imaging (DTI) and positron emission tomography (PET) have shown promise in localizing more epileptic tubers. DTI is an MRI technique that provides information regarding both the magnitude and the direction of water diffusion within a voxel. This is helpful in TSC patients because cortical and subcortical tubers have abnormal cortical architecture and altered axonal integrity, properties that can result in higher apparent diffusion coefficients (ADC) and lower fractional anisotropy (FA) in the lesions compared to normal white matter [43]. Tubers with significantly higher ADCs and lower FA have increased epileptogenic potential compared to other lesions [44, 45]. Thus DTI provides incremental information beyond morphologic MRI findings alone. Similarly, large areas of FDG-PET hypometabolism within tubers correlate with increased epileptogenicity more so than tumor size alone [46]. PET using alpha-[11C]methyl-l-tryptophan (AMT) is also helpful in identifying epileptogenic foci in TSC [45]. Tubers that have increased AMT uptake are more epileptogenic than tubers without increased AMT uptake; selective resection of such lesions improves the success of epilepsy surgery [47]. Magnetoencephalography also shows promise in improving precise localization of epileptic foci for surgical resection in TSC patients [48, 49].
White matter abnormalities
White matter abnormalities of TSC consist of radial glial bands and periventricular cyst-like lesions (Fig. 2). Radial glial bands extend from the ependymal surface of the ventricle to the cortex, sometimes terminating in a subcortical tuber [50]. Although the lesions are classically linear and referred to as bands, they can also be wedge-shaped and even tumefactive [51]. The bands are present in more than 80% of TSC patients and represent heterotopic neuronal and glial elements that arrested during cortical migration [51]. CT usually fails to demonstrate these lesions, but they are very well seen by MRI. They have similar signal characteristics to cortical tubers, with low signal on T1-W images and high signal on T2-W images. The lesions are best seen with FLAIR and rarely enhance [51, 52]. Small white matter cysts are seen in almost half of TSC patients, and their etiology is unknown [53]. They are low attenuation on CT images and isointense to CSF on all MRI pulse sequences; these lesions do not enhance [53].

White matter abnormalities and SENs in TSC. a Coronal T2-W image shows abnormal increased signal extending from the ependymal surface of the ventricle through the white matter of the right frontal lobe (white arrow). This is a radial glial band and is in continuity with a subcortical tuber in the frontal lobe (upper black arrow). Another tuber is present in the ipsilateral temporal lobe (lower black arrow). White matter cysts in the left periventricular white matter (black arrows) follow CSF signal intensity on all sequences, where they are bright on T2-W images (b) and suppressed on FLAIR images (c). SENs (white arrows) are also present, dotting the ependymal surface of the ventricles. These are partially calcified, causing some signal loss on the MR images. d Calcification within SENs is better seen on CT images as high-attenuation nodules along the ventricular surface in another patient
Subependymal nodules
Subependymal nodules (SENs) are hamartomatous lesions that dot the ependymal surface of the lateral ventricles (Fig. 2). The lesions are isointense to gray matter on MR images. SENs calcify in 90% of cases and therefore bloom secondary to dephasing artefact on MR images and are easily seen on CT images. It has been suggested that CT is useful in identifying SENs in undiagnosed family members with subtle symptoms of TSC [37].
Subependymal giant cell astrocytomas
SEGAs are low-grade neoplasms that arise near the foramen of Monro in 10–15% of patients with TSC [37] (Fig. 3). They have a mixed glioneuronal lineage [54] and some investigators have reported subtle genetic changes that might account for their growth. These findings suggest that classifying SEGAs as astrocytomas is not completely correct [55]. Nevertheless, on imaging these lesions appear similar to SENs; consideration that one is dealing with a SEGA should be made if the lesion is greater than 5 mm, incompletely calcified, or enhances [56]. Because almost all of these lesions enhance, for practical purposes, SEGAs are diagnosed by size greater than 10 mm, demonstrable growth, or the production of symptoms of hydrocephalus. A lesion is termed a “probable SEGA” if it could cause obstruction based on its size or location [55]. Suspected SEGAs should be followed by MRI at least every year [56, 57] because hydrocephalus has been reported in 40% of TSC patients with SEGAs [56].

Enlarging SEGA in a child with headache. a Axial enhanced T1-W image shows two small enhancing masses at the bilateral foramina of Monro, left (white arrow) larger than right (black arrow). b Enhanced T1-W image obtained 10 months later shows interval growth of the left SEGA (white arrow) causing mild ventricular enlargement. Difference in slice angle causes the right SEGA to appear smaller and reveals another enhancing lesion more posterior on the right (black arrows)
Abdominal manifestations and imaging findings
Renal complications are the second most common cause of morbidity and mortality in TSC patients [30]. The development of angiomyolipomas (AMLs) is very common in TSC and the formation of renal cyst is a prominent feature of TSC2 mutations. This latter feature relates to the importance of tuberin in the localization of the gene product of the autosomal-dominant polycystic kidney disease gene, PKD1 [58]; TSC2 and PKD1 are at adjacent loci on the short arm of chromosome 16 [12, 59–61]. Some have also reported an increased risk of renal cell carcinoma (RCC) [60]. Less than 10% of children with TSC have renal complications, but as they mature, they are at increased risk of renal failure and hypertension secondary to a mass effect by AMLs and cysts [30, 62]. AMLs can also bleed, causing life-threatening hemorrhage. Imaging is an excellent way to monitor for these complications, although renal US examinations in most children with TSC do not become abnormal until 7–10 years of age (55% by 7 years, 80% by 10.5 years; mean age of abnormal US examination 7.2 years) [63]. There are, of course, exceptions, as renal cysts and AMLs have been documented as early as 16 days and 4 months, respectively. For practical purposes, screening is not generally necessary until late in the first decade [62].
Angiomyolipomas
AMLs are benign hamartomatous neoplasms variably composed of smooth muscle, fat, and abnormal blood vessels [64]. The tumors are seen in almost 75% of children with TSC, and are usually multiple and bilateral [63]. However, 80% of patients with an AML do not have TSC [64]; these sporadic AMLs are usually single, unilateral, and occur in women. Mutations of the TSC2 gene have been found in both sporadic and TSC-associated AMLs. The tumors are thought to be influenced by estrogen and progesterone because they are rarely diagnosed before puberty in patients who do not have TSC, large AMLs are usually found in women, and they can grow rapidly during pregnancy. It is also known that the hormonal milieu influences TSC2 expression in laboratory animals [65]. The tumors are typically asymptomatic but grow with time [62, 63]. Conventional wisdom holds that AMLs more than 4 cm in diameter are at increased risk of bleeding (sensitivity 100%, specificity 38%) [66] and should be considered for treatment [67]. However, more recently authors have suggested that embolization of AMLs in TSC be reserved for symptomatic lesions [68]. Furthermore, investigators have also shown that a better predictor of AML hemorrhage is the presence of aneurysms larger than 5 mm (sensitivity 100%, specificity 86%) [66]. These aneurysms can be well seen by cross-sectional or conventional angiographic techniques [69].
The classic imaging appearance of an AML is that of a focal, well-marginated, fat-containing mass [62, 64, 70]. They are often cortical and usually multiple in TSC patients. The numerous interfaces of the fatty component causes AMLs to appear as echogenic masses on US examination, and they are more likely to shadow compared to RCC. The fatty component of the tumor is low attenuation on CT images and follows the signal intensity of subcutaneous fat on MR images. AMLs can also have an infiltrating pattern [62, 64, 70]; these infiltrating tumors have a more variable composition and their attenuation or signal characteristics depend on the relative amounts of fat, soft tissue, and vessels [62, 64, 70]. Because of the increased ratio of soft tissue and vessels, these lesions tend to enhance. Unfortunately, some AMLs can have aggressive features on both imaging and pathology; further complicating matters, imaging alone cannot completely differentiate between a lipid-poor AML and an RCC. However, the use of intravenous contrast agents has been shown to be useful in this second situation: a study of 81 patients (19 AMLs, 62 RCCs in a non-TSC population) showed that homogeneous or prolonged enhancement favor an AML over an RCC (P < 0.05). The presence of both of these findings had a 91% positive predictive value and 87% negative predictive value [71]. For practical purposes, imaging in the TSC population will demonstrate far more AMLs than RCCs, and the distinction is not usually part of the imager’s task (Fig. 4).

Renal AMLs and their complications. a High-resolution linear sonogram of the left kidney of a young child with TSC shows innumerable small echogenic masses (arrows) in the renal cortex. b Conventional angiogram of a large renal AML (large arrows) in a different patient demonstrates increased vascularity and markedly abnormal vessels containing several aneurysms, the largest of which was more than 5 mm in diameter (small arrow). c Axial nonenhanced CT image in an adult with TSC shows bilateral large, predominantly fatty AMLs in the upper renal poles (white arrows). The patient had acute flank pain and the right AML contains a fluid level from recent hemorrhage (black arrows). d Coronal T2-W image in a different TSC patient shows a heterogeneous mixed signal mass in the upper pole of the right kidney (large arrows) with extension into the superior vena cava (small arrows) confirmed at surgery. This was a pathologically confirmed AML
AMLs are not restricted to kidneys and can also be found in other abdominal organs [72], and even in the retroperitoneum [73] (Fig. 5). These lesions have the same imaging characteristics as renal AMLs [72]. Perhaps because they can be more confidently identified, reported cases of extrarenal AMLs tend to have the focal pattern with fatty attenuation on CT images and fat signal on MR images. Contrast enhancement is usually marked in the arterial phase with persistent enhancement during the portal venous phase. Color Doppler US shows punctiform or filiform vascular flow and on gray-scale imaging extrarenal AMLs have been reported as smoothly contoured and echogenic [72].

Two TSC patients with extrarenal fatty masses, presumed to be AMLs. a Axial enhanced CT image shows a heterogeneous but mostly low-attenuation mass causing a mild mass effect in the inferior vena cava (large arrow). A smaller lesion is present in the left hepatic lobe (small arrow). Several similar lesions were also present (not shown). b In another patient, axial enhanced CT image demonstrates a large, diffusely infiltrating fatty mass in the retroperitoneum (arrows). c A conventional renal angiogram (representative image of the left kidney during the nephrographic phase) is normal
Renal cysts
Benign epithelial renal cysts are seen in less than half of TSC patients [62, 63]. Renal cysts are associated with mutations of TSC2 because the TSC2 and PKD1 genes are at contiguous loci on chromosome 16. The combined TSC-polycystic kidney disease phenotype is seen in about 5% of TSC patients and confers a worse prognosis than TSC alone [74]. The cysts grow with time, and a mass effect leads to renal failure and hypertension. On US examination, they have the conventional cystic appearance of an anechoic mass with sharply circumscribed smooth walls and increased through-transmission. The rounded, well-defined borders are well-seen on both CT images and MR images and the cysts do not enhance. Unless complicated by hemorrhage, renal cysts show water attenuation on CT images and fluid signal on MR images [62] (Fig. 6).

Renal cysts in two patients with combined TSC-polycystic kidney disease. a Axial enhanced CT image shows numerous fluid-attenuation cysts in the left kidney, the largest of which has some rim calcification (large arrow). Several AMLs are also present (small arrows). b Coronal T2-W image shows innumerable bilateral renal cysts enlarging the kidneys
Renal cell cancer
The association of TSC with RCC has been considered controversial, but mounting evidence supports the concept that TSC patients are at increased risk for both RCC and malignant AMLs [60, 75–77]. The latter is quite rare, but it is known that RCC occurs earlier in TSC patients than in other patients. The mean age of patients with TSC-associated RCC is about 30 years, approximately 25 years younger than non-TSC patients with RCC [60, 76]. The literature is punctuated by many case reports of RCC forming in children and infants with TSC [60]. LOH at the TSC2 locus seems to be responsible for these RCCs, which develop in the dysplastic epithelial cysts associated with the TSC2-PKD1 genetic defect [75]. For practical purposes, though, RCC is very rare in TSC patients, while atypical AMLs are not uncommon. As discussed above, consideration of the enhancement characteristics of a suspicious lesion can help guide clinical management.
Perivascular epithelioid cell tumors
Within the same family of neoplasms as AML and LAM, the World Health Organization recognizes two very rare tumors called PEComa (Perivascular Epithelioid Cell + “oma”) and clear cell sugar tumor of the lung [78]. This family of neoplasms shares similar immunophenotypic characteristics and each tumor demonstrates perivascular epithelioid cell differentiation [79]. Clear cell sugar tumors of the lung are benign and only have a loose association with TSC [79, 80]. PEComas, however, are more clearly associated with TSC and some authors have suggested they be included in the diagnostic criteria for the disease [81, 82]. PEComas have no known normal tissue counterpart and their malignant potential is uncertain [83]; several patients have had documented metastatic spread [84]. These poorly understood neoplasms are more common in girls and young women and are typically found in the uterus or in other abdominopelvic sites [83, 85] (Fig. 7).

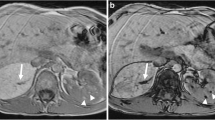
Young woman with TSC, LAM, and malignant PEComa. Axial contrast-enhanced CT image (a) and sagittal reconstructed image (b) show extensive bulky recurrence of heterogeneous masses throughout the pelvis (small white arrows) after partial surgical resection of malignant uterine PEComa. On the coronal reconstructed image (c), smaller metastatic deposits are seen in the abdomen adjacent to the right lobe of the liver (small black arrows). The patient’s native kidneys had been removed because of large AMLs causing renal failure; note the transplanted kidney in the left pelvis (long black arrow)
Cardiopulmonary manifestations and imaging findings
The most common clinically important cardiopulmonary manifestation of TSC is LAM, an unusual interstitial lung disease that causes hyperexpansion in women. Cardiac tumors are also seen in TSC, most commonly benign rhabdomyomas, but also AMLs. These are discussed in detail below.
Lymphangioleiomyomatosis
LAM is a proliferation of atypical smooth muscle cells in the axial lymphatics of the lungs that occurs almost exclusively in women [86]. Although only about 5% of TSC patients have clinically progressive lung disease, 40% of women with TSC have abnormalities seen on CT imaging. Smooth muscle proliferation and lymphatic obstruction leads to interstitial lung disease, cystic lung destruction, and chylous effusions [87]. This progressive disorder is marked by dyspnea (83%), recurrent pneumothoraces (69%), cough (66%), and hemoptysis (20%) [88, 89], and end-stage therapy requires lung transplant [90]. LAM can occur either sporadically or in association with TSC [87]; in both cases, LAM is caused by mutation of TSC2 (or, less commonly, TSC1) [91, 92]. Furthermore, 60% of patients with sporadic LAM have a primary lesion, such as leiomyoma or AML [88, 93], and genetic analysis shows identical TSC2 mutations in LAM and AML cells of these patients [91]. The pathogenesis of LAM is thought to involve a complex interplay of TSC LOH and hormonal influence [77, 91, 94]. The current pathogenetic hypothesis of the LAM centers on the concept of benign but abnormal smooth muscle cells, with TSC LOH metastasizing to the lung, usually from renal AMLs [77, 91, 94–96]. The patient’s hormonal milieu is an important factor, probably because estrogen promotes the migration of abnormal smooth muscle cells. Estrogen and progesterone receptors have been found on LAM cells [87, 97] and it is known that both LAM and AML cells are under hormonal control [98]. The recurrence of LAM after transplantation highlights and reinforces the concept of TSC mutations, benign metastasis, and hormonal environment causing LAM: recurrent LAM has been documented in previously normal (non-LAM) lungs from a male donor. In these cases, the recurrent LAM cells had identical mutations to pretransplant samples, indicating that post-transplant migration of TSC-altered cells metastasized from the recipient’s AMLs caused recurrent LAM in the transplanted lungs.
Imaging of LAM demonstrates the fairly unusual appearance of interstitial lung disease associated with hyperexpansion on chest radiographs (Fig. 8). Fine reticular opacities are seen in two-thirds of patients, and more than half of patients have large lung volumes. Cysts can be seen in about one-third of chest radiographs in LAM [99]. CT is more sensitive for LAM, and cysts are the predominant feature, seen in 100% of LAM patients. The cyst walls should be barely perceptible (but can be up to 2 mm thick) and are surrounded by normal lung [77]. The distribution is diffuse in 50% of patients, but 40% have apical sparing and the remainder have basal sparing [89]. Several investigators have reported lymphadenopathy associated with LAM [89, 99], but this was not a feature in a large series from the Armed Forces Institute of Pathology [99]. Small, noncalcified lung nodules have also been reported as a feature of LAM [88]. Pleural fluid collections are seen on CT imaging in 10–40% of LAM patients. If the fluid is chylous in nature, the attenuation should theoretically be less than zero Hounsfield units, but because they often contain increased protein content, this is often not the case [99]. LAM can also occur at extrapulmonary sites: LAM cells have been found in lymph nodes throughout the body, in the liver, and in the pancreas. Extrapulmonary LAM can be associated with chylous ascites and uterine leiomyomas [100].

LAM. a Chest radiograph demonstrates large lung volumes and diffuse fine reticular opacities throughout both lungs. Hyperexpansion in the setting of interstitial lung disease is unusual and should suggest the diagnosis of LAM, and therefore, at least consideration of TSC. b High-resolution chest CT image in a woman with TSC. Several smooth-wall cysts are present (black arrows) throughout the lungs. TSC/LAM is also associated with multifocal micronodular pneumocyte hyperplasia and a hamartomatous overgrowth of type 2 pneumocytes, and 2- to 5-mm nodules (white arrows) can often be found in these patients. c, d High-resolution chest CT with coned-down images in lung (c) and soft-tissue (d) windows shows a low-density (−5.3 HU) left pleural fluid collection, presumably a chylous effusion, a common complication of LAM
Cardiac rhabdomyomas
Cardiac rhabdomyomas are benign hamartomas and are the most common cardiac tumor in children [101, 102]. Although these tumors can be sporadic, 80% of children with rhabdomyomas are found to have TSC [103, 104]. Up to two-thirds of newborns with TSC have rhabdomyomas and they are often multiple [34]. The tumors are composed of vacuolated cardiac myocytes and are usually asymptomatic, regressing in the first year of life [7, 104, 105]. Occasionally, rhabdomyomas cause conduction abnormalities, outflow obstruction, and cardiomyopathy, and in these patients, surgical management has been advocated. Otherwise, these masses are managed conservatively [34, 103, 104]. Rhabdomyomas can be visualized by prenatal imaging as early as 22–28 weeks gestational age [102] (Fig. 9). They can be intramural but are usually intraluminal masses that protrude into ventricles. On US examination they are echogenic, and on fetal MRI they are isointense to cardiac muscle on T1-W images and hyperintense on T2-W images. Multiple small lesions can cause diffuse myocardial thickening [102]. Rhabdomyomas must be differentiated from the second most common cardiac tumor in children, ventricular fibromas [106–108]. Whereas rhabdomyomas are associated with TSC, fibromas are seen with increased frequency in children with orofacial abnormalities [109] and Gorlin (basal cell nevus) syndrome [102]; stigmata of these respective syndromes can aid in differentiating the tumors. In contrast to rhabdomyomas, fibromas tend to be solitary [102, 106]. On imaging, the diagnosis of fibroma is suggested by foci of calcification, cystic degeneration, and heterogeneous enhancement. On MR images, fibromas are usually T2 hypointense, reflecting their fibrous histology [102, 109].

Rhabdomyomas imaged during the second trimester and postnatally. a Axial T2-W fetal MR image shows several high-signal cardiac masses (arrows) in a fetus with a family history of TSC. b Postnatal cardiac MR image (four-chamber view on the left, short-axis view on the right) shows the same lesions (arrows), presumed rhabdomyomas that were later followed with echocardiography. c Contrast-enhanced CT image through the heart shows a small fat-containing mass in the left ventricle that was presumed to be a cardiac AML
Cardiac AMLs
Although thought to be quite rare, pathologically confirmed cardiac AMLs have been reported in patients not known to have TSC [110, 111]. Others have reported TSC patients with fat-containing masses that were presumed to be cardiac AMLs [112] (Fig. 9). Obviously, a conservative approach in these patients precludes differentiating lipoma from AML, both benign lesions.
Skeletal manifestations and imaging findings
The most common skeletal abnormalities of TSC are localized areas of sclerosis, commonly seen in the ribs and vertebral bodies and along the iliac side of the sacroiliac joints. The lesions are of unclear etiology but are probably hamartomas and are seen in almost all TSC patients [113]. They appear as ovoid sclerotic lesions with poorly defined margins (Fig. 10) and in today’s practice are most commonly found in vertebral bodies on CT images obtained to evaluate renal or lung disease. Radiographs of the hands and feet can show cysts in the phalanges and, occasionally, pressure erosions from periungual fibromas [113, 114] (Fig. 10).

Skeletal hamartomas. a, b Axial CT images in bone windows shows ill-defined sclerotic lesions in characteristic locations (arrows). c Coned-down radiograph of a phalanx shows a pressure erosion (arrow) from a periungual fibroma in a child with TSC
Vascular manifestations and imaging findings
Although not routinely considered part of TSC, there is a well-documented association of vascular dysplasia in this disease [115–117]. The risk of bleeding from an AML is well known and is discussed above, but the literature also contains numerous case reports describing stenosis, occlusion, and life-threatening large-vessel aneurysms that are often missed clinically [115, 117–120]. Intracranial aneurysms are the most common TSC-associated vascular anomaly outside AMLs and are best seen on T2-W or intermediate-weighted images as vascular flow voids or by MR angiography [121–125]. Aneurysms of the thoracic (TAA) and abdominal aorta (AAA) as well as of the pulmonary and brachial arteries have also been reported. AAAs have been reported in patients as young as 4.5 months; the mean age of TSC patients with AAA is 5 years, and with TAA is 11.7 years [115]. The vascular anomalies in TSC are thought to be caused by vascular hamartomas disrupting the vasa vasorum, possibly causing degeneration or dysplasia of the elastic fibers [115, 116] (Fig. 11).

Aneurysms associated with TSC. a A 3-D surface-rendered image from a CT angiogram shows a large aneurysm off the left vertebral artery (arrow) that was later coiled. b T2-W image through the circle of Willis demonstrates bilateral fusiform aneurysms of the middle cerebral arteries. These are very well seen on T2-W and proton density sequences. c, d Axial (c) and coronal (d) reformatted images from a CT angiogram reveal a massive aortic aneurysm in a 16-month-old child with TSC. Note: Images 11c & 11d courtesy of Gary Hedlund, DO
Medical imaging in management and therapy
Diagnosis
Medical imaging plays a crucial role in the diagnosis and management of TSC. The diagnostic criteria discussed above rely heavily upon imaging (Table 1). Infants and children who present with seizures and cutaneous manifestations of TSC promptly undergo brain imaging, usually with MRI. This examination alone can make a definite or probable diagnosis of TSC if any two of the following lesions are found: tuber, SEN, SEGA, and radial glial band. Even before birth, the presence of a cardiac mass alerts the physician to the possible diagnosis of TSC, allowing timely diagnosis and screening of family members. Echocardiography is also recommended in children with suspected TSC to search for rhabdomyomas. Renal imaging is helpful in confirming the diagnosis in older children and adults by demonstrating AMLs and renal cysts. Likewise, post-menarchal females with suspected TSC can undergo a high-resolution chest CT scan to identify subclinical LAM [3].
Follow-up
Patients with TSC should undergo follow-up brain imaging every 1 to 3 years, especially if any lesions have been shown to have grown [3, 10]. The presumed diagnosis of SEGA merits particularly close follow-up, and these children should be imaged each year until age 21 years (then every 2–3 years) [3, 10, 56]. The inspection of vascular flow voids should be incorporated into the radiologist’s search pattern in TSC, because intracranial aneurysms are a rare but important part of this disorder. Surveillance abdominal imaging should consist of a baseline US examination at diagnosis and serial follow-up beginning in the late first decade [3, 10, 62]. Although these scans are often limited to the kidneys, the astute imager should also briefly inspect the aorta, as there is an increased risk of AAA even in children with TSC. Similarly, a chest CT scan is often obtained to evaluate LAM in young women with LAM, and a search for aneurysms of the thoracic aorta or pulmonary artery should be made. Most experts recommend that women with LAM undergo a chest CT scan at least every 3 years. Although cardiac rhabdomyomas are asymptomatic, serial echocardiography should be performed until they involute or are shown to remain stable in size. Table 2 condenses the recommendations for diagnosis and follow-up in TSC patients.
Therapy
Therapeutic options for TSC patients revolve mainly around symptomatic management. Vigabatrin is an irreversible GABA inhibitor that has been shown to quell infantile spasms in more than 90% of TSC patients (although it is not FDA-approved for this purpose) [7]. Older children with particularly epileptogenic focal tubers have been treated by surgical resection [7]. Rapamycin is an immunosuppressant derived from bacteria that inhibits the mTOR pathway [126] and has been shown to cause regression of SEGAs [127]. It also shows promise in the treatment of AMLs and LAM [128–130]. LAM has also been treated with antiestrogen therapies, such as oophorectomy, progesterone, and tamoxifen. Lung transplantation is reserved for end-stage disease. The radiologist should be familiar with these and future treatments because successful treatment will almost certainly change the follow-up imaging [130]. Even subtle changes in a SEGA or AML or less severity of LAM could have important consequences on the patient’s clinical outcome. Of course imaging also plays an important role after tuber resection or lung transplantation. Interventional radiology plays a more direct role in the treatment of AMLs by embolization. Embolization of these tumors (with coils or sclerosing agents) should be considered when they contain aneurysmal vessels larger than 5 mm [66] and when they actively bleed. Embolization has been shown to decrease hemorrhage and preserve renal function and can be repeated if needed [7].
Conclusion
The understanding of TSC pathogenesis and genetics continues to grow, and despite available genetic testing, medical imaging continues to play an important role in its diagnosis. This role will probably expand as more and more subtle cases of TSC are discovered, and certainly, as more patients undergo treatment, imaging will play an even more important role. TSC characteristically involves multiple disparate organ systems, and therefore patients are often followed by several subspecialists, often leaving the radiologist as the only common consultant. This underscores the need for medical imagers to appreciate the depth and breadth of imaging abnormalities in this fascinating disease as well as to understand the clinical management of TSC patients. Table 3 provides a brief guide to the clinical and imaging findings discussed in this review.
References
Hurst JS, Wilcoski S (2000) Recognizing an index case of tuberous sclerosis. Am Fam Physician 61:703–708, 710
Sybert VP, Hall JG (1979) Inheritance of tuberous sclerosis. Lancet 1:783
Crino PB, Nathanson KL, Henske EP (2006) The tuberous sclerosis complex. N Engl J Med 355:1345–1356
Webb DW, Clarke A, Fryer A et al (1996) The cutaneous features of tuberous sclerosis: a population study. Br J Dermatol 135:1–5
Prather P, de Vries PJ (2004) Behavioral and cognitive aspects of tuberous sclerosis complex. J Child Neurol 19:666–674
van Baal JG, Fleury P, Brummelkamp WH (1989) Tuberous sclerosis and the relation with renal angiomyolipoma. A genetic study on the clinical aspects. Clin Genet 35:167–173
Maria BL, Deidrick KM, Roach ES et al (2004) Tuberous sclerosis complex: pathogenesis, diagnosis, strategies, therapies, and future research directions. J Child Neurol 19:632–642
Jansen FE, van Nieuwenhuizen O, van Huffelen AC (2004) Tuberous sclerosis complex and its founders. J Neurol Neurosurg Psychiatry 75:770
Pringle JJ (1890) A case of congenital adenoma sebaceum. Br J Dermatol 2:1–14
National Institute of Neurological Disorders and Stroke (2007) Tuberous sclerosis fact sheet. NIH publication no. 02-1846. National Institutes of Health, Bethesda, MD
Gomez MR, Sampson JR, Whittemore VH (1999) Tuberous sclerosis complex. Oxford University Press, New York
European Chromosome 16 Tuberous Sclerosis Consortium (1993) Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell 75:1305–1315
van Slegtenhorst M, de Hoogt R, Hermans C et al (1997) Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science 277:805–808
Knudson AG Jr (1971) Mutation and cancer: statistical study of retinoblastoma. Proc Natl Acad Sci USA 68:820–823
Ess KC (2006) The neurobiology of tuberous sclerosis complex. Semin Pediatr Neurol 13:37–42
Green AJ, Smith M, Yates JRW (1994) Loss of heterozygosity on chromosome 16p13.3 in hamartomas from tuberous sclerosis patients. Nat Genet 6:193–196
Henske EP, Scheithauer BW, Short MP et al (1996) Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am J Hum Genet 59:400–406
Sepp T, Yates JR, Green AJ (1996) Loss of heterozygosity in tuberous sclerosis hamartomas. J Med Genet 33:962–964
Verhoef S, Vrtel R, van Essen T et al (1995) Somatic mosaicism and clinical variation in tuberous sclerosis complex. Lancet 345:202
Verhoef S, Bakker L, Tempelaars AM et al (1999) High rate of mosaicism in tuberous sclerosis complex. Am J Hum Genet 64:1632–1637
Rose VM, Au KS, Pollom G et al (1999) Germ-line mosaicism in tuberous sclerosis: how common? Am J Hum Genet 64:986–992
Chan JA, Zhang H, Roberts PS et al (2004) Pathogenesis of tuberous sclerosis subependymal giant cell astrocytomas: biallelic inactivation of TSC1 or TSC2 leads to mTOR activation. J Neuropathol Exp Neurol 63:1236–1242
Dabora SL, Jozwiak S, Franz DN et al (2001) Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet 68:64–80
Jones AC, Shyamsundar MM, Thomas MW et al (1999) Comprehensive mutation analysis of TSC1 and TSC2 and phenotypic correlations in 150 families with tuberous sclerosis. Am J Hum Genet 64:1305–1315
Sancak O, Nellist M, Goedbloed M et al (2005) Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: genotype-phenotype correlations and comparison of diagnostic DNA techniques in tuberous sclerosis complex. Eur J Hum Genet 13:731–741
Jozwiak J, Jozwiak S, Wlodarski P (2008) Possible mechanisms of disease development in tuberous sclerosis. Lancet Oncol 9:73–79
Ma L, Chen Z, Erdjument-Bromage H et al (2005) Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121:179–193
Jozwiak J, Grajkowska W, Kotulska K et al (2007) Brain tumor formation in tuberous sclerosis depends on Erk activation. Neuromolecular Med 9:117–127
Ma L, Teruya-Feldstein J, Bonner P et al (2007) Identification of S664 TSC2 phosphorylation as a marker for extracellular signal-regulated kinase mediated mTOR activation in tuberous sclerosis and human cancer. Cancer Res 67:7106–7112
Online Mendelian Inheritance in Man (OMIM) (2008) #191100. Tuberous sclerosis. Johns Hopkins University, Baltimore, MD. http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=191100. Cited 3-Mar-2008
Jozwiak S, Schwartz RA, Janniger CK et al (1998) Skin lesions in children with tuberous sclerosis complex: their prevalence, natural course, and diagnostic significance. Int J Dermatol 37:911–917
Sun XF, Yan CL, Fang L et al (2005) Cutaneous lesions and visceral involvement of tuberous sclerosis. Chin Med J (Engl) 118:215–219
Sanchez NP, Wick MR, Perry HO (1981) Adenoma sebaceum of Pringle: a clinicopathologic review, with a discussion of related pathologic entities. J Cutan Pathol 8:395–403
Roach ES, Sparagana SP (2004) Diagnosis of tuberous sclerosis complex. J Child Neurol 19:643–649
Rosser T, Panigrahy A, McClintock W (2006) The diverse clinical manifestations of tuberous sclerosis complex: a review. Semin Pediatr Neurol 13:27–36
Wiznitzer M (2004) Autism and tuberous sclerosis. J Child Neurol 19:675–679
Altman NR, Purser RK, Post MJ (1988) Tuberous sclerosis: characteristics at CT and MR imaging. Radiology 167:527–532
Nixon JR, Houser OW, Gomez MR et al (1989) Cerebral tuberous sclerosis: MR imaging. Radiology 170:869–873
Baron Y, Barkovich AJ (1999) MR imaging of tuberous sclerosis in neonates and young infants. AJNR 20:907–916
Jurkiewicz E, Jozwiak S, Bekiesinska-Figatowska M et al (2006) Cyst-like cortical tubers in patients with tuberous sclerosis complex: MR imaging with the FLAIR sequence. Pediatr Radiol 36:498–501
Mizuno S, Takahashi Y, Kato Z et al (2000) Magnetic resonance spectroscopy of tubers in patients with tuberous sclerosis. Acta Neurol Scand 102:175–178
Mukonoweshuro W, Wilkinson ID, Griffiths PD (2001) Proton MR spectroscopy of cortical tubers in adults with tuberous sclerosis complex. AJNR 22:1920–1925
Karadag D, Mentzel HJ, Gullmar D et al (2005) Diffusion tensor imaging in children and adolescents with tuberous sclerosis. Pediatr Radiol 35:980–983
Jansen FE, Braun KP, van Nieuwenhuizen O et al (2003) Diffusion-weighted magnetic resonance imaging and identification of the epileptogenic tuber in patients with tuberous sclerosis. Arch Neurol 60:1580–1584
Luat AF, Makki M, Chugani HT (2007) Neuroimaging in tuberous sclerosis complex. Curr Opin Neurol 20:142–150
Chandra PS, Salamon N, Huang J et al (2006) FDG-PET/MRI coregistration and diffusion-tensor imaging distinguish epileptogenic tubers and cortex in patients with tuberous sclerosis complex: a preliminary report. Epilepsia 47:1543–1549
Kagawa K, Chugani DC, Asano E et al (2005) Epilepsy surgery outcome in children with tuberous sclerosis complex evaluated with alpha-[11C]methyl-L-tryptophan positron emission tomography (PET). J Child Neurol 20:429–438
Kamimura T, Tohyama J, Oishi M et al (2006) Magnetoencephalography in patients with tuberous sclerosis and localization-related epilepsy. Epilepsia 47:991–997
Iida K, Otsubo H, Mohamed IS et al (2005) Characterizing magnetoencephalographic spike sources in children with tuberous sclerosis complex. Epilepsia 46:1510–1517
Iwasaki S, Nakagawa H, Kichikawa K et al (1990) MR and CT of tuberous sclerosis: linear abnormalities in the cerebral white matter. AJNR 11:1029–1034
Braffman BH, Bilaniuk LT, Naidich TP et al (1992) MR imaging of tuberous sclerosis: pathogenesis of this phakomatosis, use of gadopentetate dimeglumine, and literature review. Radiology 183:227–238
Bernauer TA (1999) The radial bands sign. Radiology 212:761–762
Van Tassel P, Cure JK, Holden KR (1997) Cystlike white matter lesions in tuberous sclerosis. AJNR 18:1367–1373
Kim SK, Wang KC, Cho BK et al (2001) Biological behavior and tumorigenesis of subependymal giant cell astrocytomas. J Neurooncol 52:217–225
Goh S, Butler W, Thiele EA (2004) Subependymal giant cell tumors in tuberous sclerosis complex. Neurology 63:1457–1461
Clarke MJ, Foy AB, Wetjen N et al (2006) Imaging characteristics and growth of subependymal giant cell astrocytomas. Neurosurg Focus 20:E5
Nabbout R, Santos M, Rolland Y et al (1999) Early diagnosis of subependymal giant cell astrocytoma in children with tuberous sclerosis. J Neurol Neurosurg Psychiatry 66:370–375
Kleymenova E, Ibraghimov-Beskrovnaya O, Kugoh H et al (2001) Tuberin-dependent membrane localization of polycystin-1: a functional link between polycystic kidney disease and the TSC2 tumor suppressor gene. Mol Cell 7:823–832
Culty T, Molinie V, Lebret T et al (2006) TSC2/PKD1 contiguous gene syndrome in an adult. Minerva Urol Nefrol 58:351–354
Henske EP (2005) Tuberous sclerosis and the kidney: from mesenchyme to epithelium, and beyond. Pediatr Nephrol 20:854–857
Reeders ST, Breuning MH, Davies KE et al (1985) A highly polymorphic DNA marker linked to adult polycystic kidney disease on chromosome 16. Nature 317:542–544
Casper KA, Donnelly LF, Chen B et al (2002) Tuberous sclerosis complex: renal imaging findings. Radiology 225:451–456
Ewalt DH, Diamond N, Rees C et al (2005) Long-term outcome of transcatheter embolization of renal angiomyolipomas due to tuberous sclerosis complex. J Urol 174:1764–1766
Wagner BJ, Wong-You-Cheong JJ, Davis CJ Jr (1997) Adult renal hamartomas. Radiographics 17:155–169
Cook JD, Davis BJ, Cai SL et al (2005) Interaction between genetic susceptibility and early-life environmental exposure determines tumor-suppressor-gene penetrance. Proc Natl Acad Sci USA 102:8644–8649
Yamakado K, Tanaka N, Nakagawa T et al (2002) Renal angiomyolipoma: relationships between tumor size, aneurysm formation, and rupture. Radiology 225:78–82
Dickinson M, Ruckle H, Beaghler M et al (1998) Renal angiomyolipoma: optimal treatment based on size and symptoms. Clin Nephrol 49:281–286
Harabayashi T, Shinohara N, Katano H et al (2004) Management of renal angiomyolipomas associated with tuberous sclerosis complex. J Urol 171:102–105
Kang M, Khandelwal N, Lal A et al (2007) CT angiography in renal angiomyolipomas. Abdom Imaging 32:772–774
Logue LG, Acker RE, Sienko AE (2003) Best cases from the AFIP: angiomyolipomas in tuberous sclerosis. Radiographics 23:241–246
Kim JK, Park SY, Shon JH et al (2004) Angiomyolipoma with minimal fat: differentiation from renal cell carcinoma at biphasic helical CT. Radiology 230:677–684
Ren N, Qin LX, Tang ZY et al (2003) Diagnosis and treatment of hepatic angiomyolipoma in 26 cases. World J Gastroenterol 9:1856–1858
Tseng CA, Pan YS, Su YC et al (2004) Extrarenal retroperitoneal angiomyolipoma: case report and review of the literature. Abdom Imaging 29:721–723
Kandt RS, Haines JL, Smith M et al (1992) Linkage of an important gene locus for tuberous sclerosis to a chromosome 16 marker for polycystic kidney disease. Nat Genet 2:37–41
Al-Saleem T, Wessner LL, Scheithauer BW et al (1998) Malignant tumors of the kidney, brain, and soft tissues in children and young adults with the tuberous sclerosis complex. Cancer 83:2208–2216
Bjornsson J, Short MP, Kwiatkowski DJ et al (1996) Tuberous sclerosis-associated renal cell carcinoma. Clinical, pathological, and genetic features. Am J Pathol 149:1201–1208
Henske EP (2004) The genetic basis of kidney cancer: why is tuberous sclerosis complex often overlooked? Curr Mol Med 4:825–831
Fletcher CDM, Unni KK, Mertens F et al (2002) Pathology and genetics of tumours of soft tissue and bone. World Health Organization classification of tumours. IARC Press, Lyon, p 427
Hornick JL, Fletcher CD (2006) PEComa: what do we know so far? Histopathology 48:75–82
Flieder DB, Travis WD (1997) Clear cell “sugar” tumor of the lung: association with lymphangioleiomyomatosis and multifocal micronodular pneumocyte hyperplasia in a patient with tuberous sclerosis. Am J Surg Pathol 21:1242–1247
Pan CC, Chung MY, Ng KF et al (2007) Constant allelic alteration on chromosome 16p (TSC2 gene) in perivascular epithelioid cell tumour (PEComa): genetic evidence for the relationship of PEComa with angiomyolipoma. J Pathol 214:387–393
Bonetti F, Martignoni G, Colato C et al (2001) Abdominopelvic sarcoma of perivascular epithelioid cells. Report of four cases in young women, one with tuberous sclerosis. Mod Pathol 14:563–568
Folpe AL, Mentzel T, Lehr HA et al (2005) Perivascular epithelioid cell neoplasms of soft tissue and gynecologic origin: a clinicopathologic study of 26 cases and review of the literature. Am J Surg Pathol 29:1558–1575
Armah HB, Parwani AV (2007) Malignant perivascular epithelioid cell tumor (PEComa) of the uterus with late renal and pulmonary metastases: a case report with review of the literature. Diagn Pathol 2:45
Folpe AL, Goodman ZD, Ishak KG et al (2000) Clear cell myomelanocytic tumor of the falciform ligament/ligamentum teres: a novel member of the perivascular epithelioid clear cell family of tumors with a predilection for children and young adults. Am J Surg Pathol 24:1239–1246
Hancock E, Osborne J (2002) Lymphangioleiomyomatosis: a review of the literature. Respir Med 96:1–6
Hancock E, Tomkins S, Sampson J et al (2002) Lymphangioleiomyomatosis and tuberous sclerosis. Respir Med 96:7–13
Avila NA, Kelly JA, Chu SC et al (2000) Lymphangioleiomyomatosis: abdominopelvic CT and US findings. Radiology 216:147–153
Sherrier RH, Chiles C, Roggli V (1989) Pulmonary lymphangioleiomyomatosis: CT findings. AJR 153:937–940
Boehler A, Speich R, Russi EW et al (1996) Lung transplantation for lymphangioleiomyomatosis. N Engl J Med 335:1275–1280
Carsillo T, Astrinidis A, Henske EP (2000) Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci USA 97:6085–6090
Sato T, Seyama K, Fujii H et al (2002) Mutation analysis of the TSC1 and TSC2 genes in Japanese patients with pulmonary lymphangioleiomyomatosis. J Hum Genet 47:20–28
Avila NA, Dwyer AJ, Rabel A et al (2007) Sporadic lymphangioleiomyomatosis and tuberous sclerosis complex with lymphangioleiomyomatosis: comparison of CT features. Radiology 242:277–285
Henske EP (2003) Metastasis of benign tumor cells in tuberous sclerosis complex. Genes Chromosomes Cancer 38:376–381
Karbowniczek M, Astrinidis A, Balsara BR et al (2003) Recurrent lymphangiomyomatosis after transplantation: genetic analyses reveal a metastatic mechanism. Am J Respir Crit Care Med 167:976–982
Yu J, Astrinidis A, Henske EP (2001) Chromosome 16 loss of heterozygosity in tuberous sclerosis and sporadic lymphangiomyomatosis. Am J Respir Crit Care Med 164:1537–1540
Strizheva GD, Carsillo T, Kruger WD et al (2001) The spectrum of mutations in TSC1 and TSC2 in women with tuberous sclerosis and lymphangiomyomatosis. Am J Respir Crit Care Med 163:253–258
Logginidou H, Ao X, Russo I et al (2000) Frequent estrogen and progesterone receptor immunoreactivity in renal angiomyolipomas from women with pulmonary lymphangioleiomyomatosis. Chest 117:25–30
Abbott GF, Rosado-de-Christenson ML, Frazier AA et al (2005) From the archives of the AFIP: lymphangioleiomyomatosis: radiologic-pathologic correlation. Radiographics 25:803–828
Torres VE, Bjornsson J, King BF et al (1995) Extrapulmonary lymphangioleiomyomatosis and lymphangiomatous cysts in tuberous sclerosis complex. Mayo Clin Proc 70:641–648
Beghetti M, Gow RM, Haney I et al (1997) Pediatric primary benign cardiac tumors: a 15-year review. Am Heart J 134:1107–1114
Grebenc ML, Rosado de Christenson ML, Burke AP et al (2000) Primary cardiac and pericardial neoplasms: radiologic-pathologic correlation. Radiographics 20:1073–1103; quiz 1110–1111, 1112
Bosi G, Lintermans JP, Pellegrino PA et al (1996) The natural history of cardiac rhabdomyoma with and without tuberous sclerosis. Acta Paediatr 85:928–931
Carrico A, Moura C, Baptista MJ et al (2001) Cardiac rhabdomyomas presenting in neonates. Rev Port Cardiol 20:1095–1101
Jozwiak S, Kotulska K, Kasprzyk-Obara J et al (2006) Clinical and genotype studies of cardiac tumors in 154 patients with tuberous sclerosis complex. Pediatrics 118:e1146–e1151
Yu K, Liu Y, Wang H et al (2007) Epidemiological and pathological characteristics of cardiac tumors: a clinical study of 242 cases. Interact Cardiovasc Thorac Surg 6:636–639
Piazza N, Chughtai T, Toledano K et al (2004) Primary cardiac tumours: eighteen years of surgical experience on 21 patients. Can J Cardiol 20:1443–1448
Thomas-de-Montpreville V, Nottin R, Dulmet E et al (2007) Heart tumors in children and adults: clinicopathological study of 59 patients from a surgical center. Cardiovasc Pathol 16:22–28
Isaacs H Jr (2004) Fetal and neonatal cardiac tumors. Pediatr Cardiol 25:252–273
Shimizu M, Manabe T, Tazelaar HD et al (1994) Intramyocardial angiomyolipoma. Am J Surg Pathol 18:1164–1169
Tsai CC, Chou CY, Han SJ et al (1997) Cardiac angiomyolipoma: radiologic and pathologic correlation. J Formos Med Assoc 96:653–656
Winterkorn EB, Dodd JD, Inglessis I et al (2007) Tuberous sclerosis complex and myocardial fat-containing lesions: a report of four cases. Clin Genet 71:371–373
Green GJ (1968) The radiology of tuberose sclerosis. Clin Radiol 19:135–147
Holt JF, Dickerson WW (1952) The osseous lesions of tuberous sclerosis. Radiology 58:1–8
Jost CJ, Gloviczki P, Edwards WD et al (2001) Aortic aneurysms in children and young adults with tuberous sclerosis: report of two cases and review of the literature. J Vasc Surg 33:639–642
Restrepo R, Ranson M, Chait PG et al (2003) Extracranial aneurysms in children: practical classification and correlative imaging. AJR 181:867–878
Rolfes DB, Towbin R, Bove KE (1985) Vascular dysplasia in a child with tuberous sclerosis. Pediatr Pathol 3:359–373
Burrows NJ, Johnson SR (2004) Pulmonary artery aneurysm and tuberous sclerosis. Thorax 59:86
Hagood CO Jr, Garvin DD, Lachina FM et al (1976) Abdominal aortic aneurysm and renal hamartoma in an infant with tuberous sclerosis. Surgery 79:713–715
Tamisier D, Goutiere F, Sidi D et al (1997) Abdominal aortic aneurysm in a child with tuberous sclerosis. Ann Vasc Surg 11:637–639
Beall S, Delaney P (1983) Tuberous sclerosis with intracranial aneurysm. Arch Neurol 40:826–827
Brill CB, Peyster RG, Hoover ED et al (1985) Giant intracranial aneurysm in a child with tuberous sclerosis: CT demonstration. J Comput Assist Tomogr 9:377–380
Jurkiewicz E, Jozwiak S (2006) Giant intracranial aneurysm in a 9-year-old boy with tuberous sclerosis. Pediatr Radiol 36:463
Chatrath R, Noronha PA (2000) Intracranial aneurysm in tuberous sclerosis. Int Pediatr 15:152–154
Beltramello A, Puppini G, Bricolo A et al (1999) Does the tuberous sclerosis complex include intracranial aneurysms? A case report with a review of the literature. Pediatr Radiol 29:206–211
Huang S, Bjornsti MA, Houghton PJ (2003) Rapamycins: mechanism of action and cellular resistance. Cancer Biol Ther 2:222–232
Franz DN, Leonard J, Tudor C et al (2006) Rapamycin causes regression of astrocytomas in tuberous sclerosis complex. Ann Neurol 59:490–498
Herry I, Neukirch C, Debray MP et al (2007) Dramatic effect of sirolimus on renal angiomyolipomas in a patient with tuberous sclerosis complex. Eur J Intern Med 18:76–77
Lee L, Sudentas P, Donohue B et al (2005) Efficacy of a rapamycin analog (CCI-779) and IFN-gamma in tuberous sclerosis mouse models. Genes Chromosomes Cancer 42:213–227
Bissler JJ, McCormack FX, Young LR et al (2008) Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med 358:140–151
Roach ES, Gomez MR, Northrup H (1998) Tuberous sclerosis complex consensus conference: revised clinical diagnostic criteria. J Child Neurol 13:624–628
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Baskin, H.J. The pathogenesis and imaging of the tuberous sclerosis complex. Pediatr Radiol 38, 936–952 (2008). https://doi.org/10.1007/s00247-008-0832-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00247-008-0832-y