
Abstract
Background
Atypical teratoid/rhabdoid tumor (AT/RT) is a rare aggressive infantile neoplasm of uncertain origin. This study was performed to assess the clinicopathologic and immunohistochemical features of four AT/RT cases.
Case reports
Two cases were male and two were female, and their ages ranged from 8 to 103 months. Tumors were located in the cerebellum (two cases), frontoparietal lobe (one case), and third ventricle (one case). Histopathologically, the tumors were composed of rhabdoid cells and undifferentiated small cells mixed with epithelial or mesenchymal components. However, one of the tumors was composed predominantly of a mesenchymal component mimicking a sarcoma. Immunohistochemically, vimentin (4/4), epithelial membrane antigen (4/4), cytokeratin (3/4), smooth muscle actin (4/4), glial fibrillary acidic protein (4/4), S-100 (4/4), and synaptophysin (1/4) were positive in varying proportions, while desmin and INI-1 were negative in all the cases. All of the patients died within a mean of 14 months due to tumor progression despite the chemotherapy. Only one of our patients lived for 40 months after the diagnosis. In conclusion, AT/RTs are aggressive tumors. They can occur in a variety of locations, such as the third ventricle. Morphologically, a large spectrum can be seen, like predominantly sarcoma in appearance, but immunohistochemistry is helpful in the correct diagnosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Atypical teratoid/rhabdoid tumor (AT/RT) of the central nervous system (CNS) is a distinct tumor, based on morphological, immunohistochemical, and cytogenetic features, and is usually seen in young children less than 2 years of age [1]. AT/RT was described by Rorke et al. [2] as a special type of CNS tumor characterized with rhabdoid cells, areas resembling primitive neuroectodermal tumor (PNET) and sometimes foci of malignant mesenchymal and/or epithelial tissue. Therefore, it was diagnosed as a medulloblastoma, choroid plexus carcinoma, or PNET before the definition of this tumor as a distinct entity [3]. In 1993, it was included in the World Health Organization classification as a grade IV embryonal neoplasm [4].
AT/RTs demonstrate a wide range of immunohistochemical reactivity because of the divergent differentiation of the tumor cells. The tumor cells show variable immunoreactivity for epithelial membrane antigen (EMA), cytokeratin, vimentin, smooth muscle actin (SMA), desmin, glial fibrillary acidic protein (GFAP), and synaptophysin [1, 5, 6]. In addition to these markers, INI-1 protein has been reported as a useful marker in the differential diagnosis of AT/RT from medulloblastoma/PNET, choroid plexus carcinoma, and the other brain tumors that can mimic AT/RT [7, 8]. In AT/RT, biallelic inactivation of the INI-1 gene located in 22q11.2 causes lack of expression of INI-1 protein in all the tumor cells [7, 9]. In this study, we present the clinicopathologic and immunohistochemical features of four AT/RT cases.
Case reports
Case 1
An 8-year-old boy developed ataxia and gait disturbance. His magnetic resonance imaging (MRI) scan demonstrated a mass in the posterior fossa extending through the supratentorium. He underwent a subtotal resection of the tumor and a shunt was placed. On histological examination, strands and cords of rhabdoid cells were observed in most of the slides. The rhabdoid cells had abundant eosinophilic cytoplasm and clear eccentric nucleus with a prominent nucleolus (Fig. 1). The component of PNET was only focally seen. An immunohistochemical panel was performed, and the results are summarized in Table 2. The diagnosis was AT/RT. He was started on ifosfamide, carboplatin, and etoposide (ICE) chemotherapy. The tumor progressed after the third course of this treatment, and he died of progressive disease 4 months after the diagnosis.

The tumor is composed of rhabdoid cells with prominent nucleolus and abundant eosinophilic cytoplasm (Case 1) (hematoxylin–eosin, original magnification ×200)
Case 2
A 2-year-old boy complained of vomiting for 45 days. A computerized tomography (CT) scan showed right cerebellar mass. He underwent a subtotal removal of the tumor. A second surgery after 3 months was done upon tumor enlargement during the follow-up, with near-total excision of the tumor. Histologically, the tumor consisted of rhabdoid cells and undifferentiated small cells mixed with epithelioid component (Fig. 2). The immunohistochemical results of the tumors are summarized in Table 2. Pathology revealed AT/RT. He was started on chemotherapy; however, the tumor progressed and invaded the brain stem. A third operation was done as subtotal excision. He died of disease progression 40 months after the diagnosis.

Epithelioid cells showing a few glandular components and rhabdoid cells are seen (Case 2) (hematoxylin–eosin, original magnification ×200)
Case 3
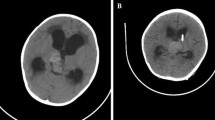
An 8-month-old female infant presented with convulsion. CT scan showed a large cystic frontoparietal mass on the left (Fig. 3). She underwent a partial resection of the tumor. Histologically, the tumor was characterized with mesenchymal appearance predominantly and rhabdoid and undifferentiated small cells focally. In this case, spindle cells were tightly arranged in a fascicular pattern resembling a sarcoma (Fig. 4a). In the focal areas, cords of cells in a mucinous background resembling a chordoma were seen (Fig. 4b). Pathological examination revealed AT/RT. She was started on ICE chemotherapy. During follow-up, her tumor progressed, and she died 5 months after the diagnosis, after the fifth chemotherapy course.

T1 (a), T2 (b) and contrast-enhanced T1-weighted (c) axial MR images of Case 3 show a highly invasive cystic mass located on the frontal and left parietal lobes. The mass effect of the tumor caused a serious midline shift, and both anterior horns of the ventricular system are compressed. Contrast-enhanced axial T1-weighted image (c) shows a strong heterogeneous enhancement within and around the tumor

a Spindle cells are tightly arranged in a fascicular pattern resembling a sarcoma. b Cords of cells in a mucinous background resembling a chordoma are seen in the focal areas (Case 3) (hematoxylin–eosin, original magnification ×400)
Case 4
A 2-year-old female patient presented with acute loss of consciousness. Her MRI scan revealed a solid mass with strong contrast enhancement filling the third ventricle. She underwent a gross total resection of the tumor. On histological examination, rhabdoid cells, undifferentiated small cells, and epithelioid components were observed. The histological features were similar to those of Case 2. The diagnosis was AT/RT. ICE chemotherapy treatment was started, but after the third course of further treatment it was refused. She died of tumor progression 6 months after the diagnosis.
The characteristics of the patients are summarized in Table 1 and the immunohistochemical results of the tumors are in Table 2.
Discussion
Atypical teratoid/rhabdoid tumor of the CNS is an aggressive tumor that primarily occurs in very young children usually less than 2 years of age [6, 10]. Rorke et al. [3] reported 52 patients with AT/RT and most of the cases were aged 3 years or younger. However, in several published series, patients over 3 years of age have been reported. Packer et al. [11] reported 42 AT/RT cases, 22 of whom were older than 3 years of age. Moreover, AT/RT may occur in adults [12, 13]. In the present study, the mean age of the patients was 39 months (range, 8 to 103 months). Three of our cases were less than 2 years of age and one case was over 2 years old at the time of diagnosis.
AT/RTs are commonly located in the cerebellum but they can arise anywhere in the CNS [3, 10, 14]. Localization of AT/RT in the third ventricle is a rare finding. Donovan et al. [15] reported a case of AT/RT located in the midline of the lateral ventricle and roof of the third ventricle. In the present study, two of the tumors were located in the cerebellum, one in the frontal lobe and one in the third ventricle.
The distinction of AT/RT from PNET/medulloblastoma is of clinical significance because the prognosis of AT/RT is poorer than of PNET/medulloblastoma despite the aggressive therapy [1, 3, 10]. The diagnosis of AT/RT and its distinction from PNET/medulloblastoma depend on histological and immunohistochemical features. On histologic examination, rhabdoid cells, which are defined as the histologic hallmark of the AT/RTs, are seen in most of the cases [2, 3]. These cells are described as large, pale cells with oval, polygonal, or elongated nuclei and eosinophilic or pale cytoplasm. The nuclei have open chromatin pattern and small to moderately prominent nucleolus [3, 6, 10]. Presence of small cells mimicking PNET/medulloblastoma is a more common feature than the mesenchymal and epithelial components in AT/RTs [3, 6, 10].
In the present study, rhabdoid cells were seen in all the cases. Of the four cases, two consisted of an undifferentiated small cell component mimicking PNET/medulloblastoma. The epithelioid component was evident in two cases. One of the cases had a sarcoma aspect characterized by atypical spindle cells with long irregular fascicles. In the focal areas, there were rhabdoid cells, small round cells, and cords of cells in a mucinous background resembling a chordoma, but the immunohistochemical findings (positivity for vimentin, EMA, cytokeratin, SMA, GFAP, and S-100; negativity for synaptophysin, desmin, and INI-1) were compatible with AT/RT.
Immunohistochemical examination is essential for distinguishing AT/RT from PNET/medulloblastoma. AT/RTs show variable immunoreactivity for EMA, vimentin, cytokeratin, GFAP, SMA, desmin, and synaptophysin [1, 3, 10]. In the present study, our immunohistochemical results were similar to the studies reported in the literature. All the cases were immunoreactive for EMA, vimentin and SMA, but they showed variable expression for cytokeratin, GFAP, S100 and synaptophysin. None of the cases was immunoreactive for desmin.
Besides the mentioned immunohistochemical markers, INI-1 protein showing negative immunoreactivity in the tumor cells of AT/RTs has been found in recent years [7, 16]. The INI-1 protein, which is a component of the mammalian SWI/SNF complex, functions in an adenosine triphosphate-dependent manner to alter the structure of chromatin, but its role in malignant transformation is unknown [17]. In several studies, INI-1 protein has been suggested as a useful marker for confirming the diagnosis of AT/RT and distinguishing it from PNET/medulloblastoma and the other tumors [7, 8, 12]. In the present study, immunohistochemically, none of the four cases was immunoreactive with INI-1 protein, similar to the previous studies reported.
AT/RTs are aggressive tumors with a dismal prognosis and there is currently no effective treatment of these tumors [1, 2]. The mean survival for AT/RT has been reported as 3 to 19 months [6, 18, 19]. However, Bouvier et al. [17] presented a case diagnosed as AT/RT with a 7-year event-free survival after surgery and cranial radiotherapy. In the present study, all of the patients died within a mean of 14 months (range, 4 to 40 months) due to progressive disease despite the chemotherapy.
In conclusion, as AT/RTs show an aggressive clinical course, they should be differentiated from the embryonal tumors with which they are often confused. Rare localizations of these tumors, such as in the third ventricle, should be kept in mind. For confirmation of the diagnosis of AT/RT, it is essential to demonstrate the negative immunoreactivity for INI-1 and the variable expression of the mentioned immunohistochemical markers. INI-1 protein is a useful marker for these tumors, particularly those that do not show evidence of rhabdoid cells, which are described as the histological hallmark of AT/RTs.
References
Oka H, Scheithauer BW (1999) Clinicopathologic characteristics of atypical teratoid/rhabdoid tumor. Neurol Med Chir 39:510–518
Rorke LB, Packer R, Biegel J (1995) Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. J Neurooncol 24:21–28
Rorke LB, Packer R, Biegel J (1996) Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood: A definition of an entity. J Neurosurg 85:56–65
Kleihues P, Louis DN, Scheithauer BW et al (2002) The WHO classification of tumors of the nervous system. J Neuropathol Exp Neurol 61:215–225
Reddy AT (2005) Atypical teratoid/rhabdoid tumors of the central nervous system. J Neurooncol 75:309–313
Bhattacharigee M, Hicks J, Langford L et al (1997) Central nervous system atypical teratoid/rhabdoid tumors of infancy and childhood. Ultrastruct Pathol 21:369–378
Judkins AR, Mauger J, Ht A, Rorke LB, Biegel JA (2004) Immunohistochemical analysis of hSNF5/INI1 in pediatric CNS neoplasms. Am J Surg Pathol 28:644–650
Judkins AR, Burger PC, Hamilton RL et al (2005) INI1 protein expression distinguishes atypical teratoid/rhabdoid tumor from choroid plexus carcinoma. J Neuropathol Exp Neurol 64:391–397
Hoot AC, Russo P, Judkins AR, Perlman EJ, Biegel JA (2004) Immunohistochemical analysis of hSNF5/INI1 distinguishes renal and extra-renal malignant rhabdoid tumors from other pediatric soft tissue tumors. Am J Surg Pathol 28:1485–1491
Burger PC, Yu IT, Tihan T et al (1998) Atypical teratoid/rhabdoid tumor of the central nervous system: a highly malignant tumor of infancy and childhood frequently mistaken for medulloblastoma: a Pediatric Oncology Group study. Am J Surg Pathol 22:1083–1092
Packer RJ, Biegel JA, Blaney S et al (2002) Atypical teratoid/rhabdoid tumor of the central nervous system: report on workshop. J Pediatr Hematol Oncol 24:337–342
Makuria AT, Rushing EJ, McGrail KM, Hartmann DP, Azumi N, Ozdemirli M (2008) Atypical teratoid rhabdoid tumor (AT/RT) in adults: review of four cases. J Neurooncol 88:321–330
Horn M, Schlote W, Lerch KD, Steudel WI, Harms D, Thomas E (1992) Malignant rhabdoid tumor: primary intracranial manifestation in an adult. Acta Neuropathol 83:445–448
Lee MC, Park SK, Lim JS et al (2002) Atypical teratoid/rhabdoid tumor of the central nervous system: clinico-pathological study. Neuropathology 22:252–260
Donovan DJ, Smith AB, Petermann GW (2006) Atypical teratoid/rhabdoid tumor of the velum interpositum presenting as a spontaneous intraventricular hemorrhage in an infant: case report with long-term survival. Pediatr Neurosurg 42:187–192
Perry A, Fuller CE, Judkins AR, Dehner LP, Biegel JA (2005) INI1 expression is retained in composite rhabdoid tumors, including rhabdoid meningiomas. Mod Pathol 18:951–958
Bouvier C, De Paula AM, Fernandez C et al (2008) Atypical teratoid/rhabdoid tumour: 7-year event-free survival with gross total resection and radiotherapy in a 7-year-old boy. Childs Nerv Syst 24:143–147
Hilden JW, Watterson J, Longee DC et al (1998) Central nervous system atypical teratoid/rhabdoid tumor: response to intensive therapy and review of the literature. J Neurooncol 40:265–275
Ho DM, Hsu CY, Wong TT, Ting LT, Chiang H (2000) Atypical teratoid/rhabdoid tumor of the central nervous system: a comparative study with primitive neuroectodermal tumor/medulloblastoma. Acta Neuropathol 99:482–488
Acknowledgment
The authors thank Dr. Tarık Tihan (Department of Pathology, University of California) for performing immunohistochemistry of the INI-1 protein.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ertan, Y., Sezak, M., Turhan, T. et al. Atypical teratoid/rhabdoid tumor of the central nervous system: clinicopathologic and immunohistochemical features of four cases. Childs Nerv Syst 25, 707–711 (2009). https://doi.org/10.1007/s00381-009-0811-0
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00381-009-0811-0