
Abstract
Lamotrigine has been used for patients with epilepsy and/or bipolar disorder, overdose of which induced the hypotension, elevation of the atrial pacing threshold, cardiac conduction delay, wide complex tachycardia, cardiac arrest and Brugada-like electrocardiographic pattern. To clarify how lamotrigine induces those cardiovascular adverse events, we simultaneously assessed its cardiohemodynamic and electrophysiological effects using the halothane-anesthetized dogs (n = 4). Lamotrigine was intravenously administered in doses of 0.1, 1 and 10 mg/kg/10 min under the monitoring of cardiovascular variables, possibly providing subtherapeutic to supratherapeutic plasma concentrations. The low or middle dose of lamotrigine did not alter any of the variables. The high dose significantly delayed the intra-atrial and intra-ventricular conductions in addition to the prolongation of ventricular effective refractory period, whereas no significant change was detected in the other variables. Lamotrigine by itself has relatively wide safety margin for cardiohemodynamics, indicating that clinically reported hypotension may not be induced through its direct action on the resistance arterioles or capacitance venules. The electrophysiological effects suggested that lamotrigine can inhibit Na+ channel in the in situ hearts. This finding may partly explain the onset mechanism of lamotrigine-associated cardiac adverse events in the clinical cases. In addition, elevation of J wave was induced in half of the animals, suggesting that lamotrigine may have some potential to unmask Brugada electrocardiographic genotype in susceptible patients.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lamotrigine has been used for patients with epilepsy and/or bipolar disorder, overdose of which caused hypotension, cardiac conduction delay, wide complex tachycardia and cardiac arrest [1]. Moreover, lamotrigine was reported to elevate the atrial pacing threshold in a patient who had been implanted a pacemaker [2], and also to induce Brugada-like electrocardiographic changes in patients with its overdose [3] as well as in the presence of comorbidity diseases such as hypertension and dyslipidemia [4] and combined medicines like class Ia antiarrhythmic drug ajmaline [5]. The inhibitory effects of lamotrigine on ionic channels in the central nervous system have been extensively studied; namely, Na+ channels in N4TG1 mouse neuroblastoma clonal cells, recombinant rat brain type IIA Na+ channels expressed in Chinese hamster ovary cells, native Na+ channels in rat hippocampal pyramidal neurons and cultured rat cerebellar granule cells, and R-type and T-type Ca2+ channels in human embryonic kidney 293 cells [6,7,8,9]. However, information regarding in vitro and in vivo cardiac effects of lamotrigine that can explain the onset mechanisms of those clinically reported, cardiovascular adverse events remains to be lacking except that it can inhibit IKr channel with IC50 of 229 µM in human embryo kidney 293 cells [10].
To clarify how lamotrigine induces those cardiovascular adverse events, we simultaneously assessed its cardiohemodynamic and electrophysiological effects using the halothane-anesthetized beagle dogs, which have been known to be able to predict the electropharmacological responses in healthy human subjects [11]. Moreover, to better perform in vivo reverse translational study, we measured the following electrophysiological variables. First, His bundle electrogram and monophasic action potential (MAP) were recorded in addition to standard lead II electrocardiogram so as to precisely analyze its effects on the depolarization and repolarization phases. Second, MAP and effective refractory period were simultaneously assessed at the same site to predict the terminal repolarization period, which can quantify the risk of a drug to induce reentrant ventricular arrhythmias [11]. Third, J wave in the electrocardiogram was monitored to analyze the magnitude of drug-induced early repolarization [12, 13].
Materials and methods
Experiments were performed in female beagle dogs weighing approximately 9 kg (n = 4). Animals were obtained through Kitayama Labes Co., Ltd. (Nagano, Japan). All experiments were approved by the Toho University Animal Care and User Committee (No. 19-52-395) and performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of Toho University. Dogs were initially anesthetized with thiopental sodium (30 mg/kg, i.v.). After intubation with a cuffed endotracheal tube, anesthesia was maintained by inhalation of halothane (1% v/v) vaporized in oxygen with a volume-limited ventilator (SN-480-3; Shinano Manufacturing Co., Ltd., Tokyo, Japan). Tidal volume and respiratory rate were set at 20 mL/kg and 15 breaths/min, respectively.
Cardiohemodynamic variables
Four clinically available catheter-sheath sets (FAST-CATH™ 406119; St. Jude Medical, Daig Division, Inc., Minnetonka, MN, USA) were used; two were inserted into the right and left femoral arteries toward abdominal aorta, the other two were done into the right and left femoral veins toward inferior vena cava, respectively. A pig-tail catheter was placed at the left ventricle to measure the left ventricular pressure through the right femoral artery, whereas the aortic pressure was measured at a space between inside of the catheter sheath and outside of the pig-tail catheter through a flush line. The maximum upstroke velocities of the left ventricular pressure and the left ventricular end-diastolic pressure were obtained during sinus rhythm to estimate the inotropic status and the preload to left ventricle, respectively. A thermodilution catheter (132F5; Edwards Lifesciences, Irvine, CA, USA) was positioned at the right side of the heart through the right femoral vein. The cardiac output was measured with a standard thermodilution method using a cardiac output computer (MFC-1100; Nihon Kohden Corporation, Tokyo, Japan). The total peripheral vascular resistance was calculated with the basic equation: total peripheral vascular resistance = mean blood pressure/cardiac output.
Electrophysiological variables
The surface lead II electrocardiogram was obtained from the limb electrodes. Corrected QT interval (QTc) was calculated with Van de Water’s formula: QTc = QT − 0.087 × (RR − 1,000) with RR given in ms [14]. J wave was defined as an elevation of the QRS-ST junction of at least 0.1 mV from baseline, manifested as QRS slurring or notching [12]. A standard 6 French quad-polar electrodes catheter (Cordis-Webster Inc., Baldwin Park, CA, USA) was positioned at the non-coronary cusp of the aortic valve through the left femoral artery to obtain the His bundle electrogram.
A bi-directional steerable MAP recording/pacing combination catheter (1675P; EP Technologies, Inc., Sunnyvale, CA, USA) was positioned at the endocardium of the right ventricle through the catheter sheath placed at the left femoral vein to obtain MAP signals. The signals were amplified with a DC preamplifier (model 300; EP Technologies, Inc.). The MAP duration (ms) at 90% repolarization level was defined as MAP90. The heart was electrically driven using a cardiac stimulator (SEC-3102; Nihon Kohden Corporation) with the pacing electrodes of the combination catheter placed in the right ventricle. The stimulation pulses were rectangular in shape 1–2 V of amplitude (about twice the threshold voltage) and of 1-ms duration. The MAP90 was measured during sinus rhythm (MAP90(sinus)) and at pacing cycle lengths of 400 ms (MAP90(CL400)) and 300 ms (MAP90(CL300)).
The effective refractory period of the right ventricle was assessed with programmed electrical stimulation. The pacing protocol consisted of 5 beats of basal stimuli in a cycle length of 400 ms followed by an extra stimulus of various coupling intervals. The duration of the terminal repolarization period of the ventricle was calculated by the difference between the MAP90(CL400) and effective refractory period at the same site, which reflects the extent of electrical vulnerability of the ventricular muscle [11].
Experimental protocol
The aortic and left ventricular pressures, electrocardiogram, His-bundle electrogram and MAP signals were monitored with a polygraph system (RM-6000; Nihon Kohden Corporation), which were analyzed with a real-time fully automatic data analysis system (WinVAS3 ver. 1.1R24; Physio-Tech Co., Ltd., Tokyo, Japan). Each measurement of electrocardiogram and MAP variables as well as atrio-His (AH) and His-ventricular (HV) intervals adopted the mean of three recordings of consecutive complexes. After the basal assessment, lamotrigine in a low dose of 0.1 mg/kg was intravenously administered over 10 min, and each variable was assessed at 5, 10, 15, 20 and 30 min after the start of infusion. Then, lamotrigine in a middle dose of 1 mg/kg was intravenously administered over 10 min, and each variable was assessed in the same manner. Finally, lamotrigine in a high dose of 10 mg/kg was intravenously administered over 10 min, and each variable was assessed at 5, 10, 15, 20, 30, 45 and 60 min after the start of infusion.
Rationale of the drug doses
According to the drug information from the manufacturer (Glaxo Smith Kline K.K., Tokyo, Japan, published November 2018, version 11), usual maintenance doses for epilepsy and bipolar disorder of lamotrigine are described as 100–200 mg/day and 200 mg/day, respectively, both of which can be elevated up to 400 mg/day. Also, the single oral administration of 200 mg of lamotrigine was shown to provide peak plasma concentration (Cmax) of 3.08 µg/mL (12 µmol/L). In our previous studies [13, 15, 16], we used the same doses and the same experimental protocol to assess cardiovascular effects of some drugs including TK159, milnacipran and fluvoxamine, and simultaneously determined the plasma drug concentrations. In those studies, intravenous infusion of 0.1, 1 and 10 mg/kg over 10 min of the 3 drugs provided Cmax values of 0.10 ± 0.02, 0.85 ± 0.29 and 9.40 ± 2.88 µg/mL at the end of infusion, respectively in the halothane-anesthetized dogs [13, 15, 16]. Using this information, we roughly estimated that lamotrigine in doses of 0.1, 1 and 10 mg/kg would provide similar Cmax values, which could be considered to reflect its subtherapeutic to supratherapeutic concentrations. As the plasma protein-binding ratio of lamotrigine was reported to be 53.1–56.2%, the low, middle and high doses could provide peak free plasma concentrations of approximately 0.04–0.05 (0.17–0.18), 0.37–0.40 (1.45–1.56) and 4.1–4.4 µg/mL (16.0–17.2 µmol/L), respectively.
Drugs
Lamotrigine (Tokyo Chemical Industry Co., Ltd., Tokyo, Japan) was dissolved with 2% lactic acid in concentrations of 0.1, 1 and 10 mg/mL. Other drugs used were thiopental sodium (Ravonal® 0.5 g for Injection, Mitsubishi Tanabe Pharma Co., Osaka, Japan), halothane (Fluothane®, Takeda Pharmaceutical Co., Ltd., Osaka, Japan) and heparin calcium (Caprocin®, Sawai Pharmaceutical Co., Ltd., Osaka, Japan).
Statistical analysis
Data are presented as mean ± SEM. Differences within a parameter were evaluated with one-way, repeated-measures analysis of variance (ANOVA) followed by contrasts as a post hoc test for mean values comparison. A p value < 0.05 was considered to be significant.
Results
No animals exerted any lethal ventricular arrhythmias or hemodynamic collapse leading to the animals’ death during the experiment.
Effects on the cardiohemodynamic variables
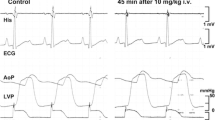
The time courses of changes in the cardiohemodynamic variables are summarized in Fig. 1. The pre-drug basal control values (C) of the heart rate, mean blood pressure, cardiac output, total peripheral vascular resistance, maximum upstroke velocity of left ventricular pressure and left ventricular end-diastolic pressure were 112 ± 7 beats/min, 121 ± 12 mmHg, 3.2 ± 0.4 L/min, 39 ± 5 mmHg·min/L, 2,592 ± 455 mmHg/s and 12 ± 1 mmHg, respectively. The low, middle or high dose did not alter any of the variables.

Time courses of changes in the heart rate (HR), mean blood pressure (MBP), cardiac output (CO), total peripheral vascular resistance (TPR), maximum upstroke velocity of the left ventricular pressure (LVdP/dtmax) and left ventricular end-diastolic pressure (LVEDP) after the administration of lamotrigine. Data are presented as mean ± SEM (n = 4)
Effects on the electrocardiographic variables
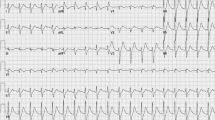
Typical tracings of the electrocardiogram are depicted in Fig. 2, and the time courses of changes in the electrocardiographic variables are summarized in Fig. 3. The pre-drug basal control values (C) of the PR interval, QRS width, QT interval, QTc and P-wave duration were 108 ± 6 ms, 64 ± 2 ms, 290 ± 15 ms, 330 ± 14 and 52 ± 2 ms, respectively. The low and middle doses did not alter any of the variables. The high dose prolonged the PR interval for 45–60 min, the QRS width at 10 and 60 min and the P-wave duration for 5–60 min after the start of infusion, whereas no significant change was detected in the other variables. J wave that satisfied its criteria was not observed before the administration of the drug, whereas it was detected in 2 out of 4 animals for 5–60 min and 30–60 min, respectively, after the high dose, the latter of which was depicted in Fig. 2.

Typical tracings showing the lead II electrocardiogram (ECG) during sinus rhythm at pre-drug basal control (Control, top) and 60 min after the start of 10 mg/kg of lamotrigine infusion (60 min after 10 mg/kg, i.v., bottom). Note the enlarged J wave after the drug administration (arrow)

Time courses of changes in the PR interval, QRS width, QT interval, QTc, P-wave duration (P duration), atrio-His (AH) and His-ventricular (HV) intervals; and the MAP90 during sinus rhythm (MAP90(sinus)), MAP90 at cycle lengths of 400 ms (MAP90(CL400)) and 300 ms (MAP90(CL300)), effective refractory period of the right ventricle (ERP(CL400)) and terminal repolarization period (TRP) after the administration of lamotrigine. QT interval was corrected with Van de Water's formula: QTc = QT − 0.087 × (RR − 1,000). MAP90: monophasic action potential duration at 90% repolarization level. Data are presented as mean ± SEM (n = 4). Closed symbols represent significant differences from the corresponding pre-drug basal control value (C) by p < 0.05
Effects on the AH and HV intervals, and MAP90 (sinus) during sinus rhythm
The time courses of changes in the AH and HV intervals, and MAP90 during sinus rhythm are summarized in Fig. 3. Their pre-drug basal control values (C) were 73 ± 7, 22 ± 2 and 247 ± 4 ms, respectively. The low or middle dose did not alter any of the variables. The high dose prolonged the HV interval at 15 min and for 45–60 min, whereas no significant change was detected in the other variables.
Effects on the MAP90 (CL400), MAP90 (CL300), effective refractory period and terminal repolarization period
The time courses of changes in the MAP90(CL400), MAP90(CL300), effective refractory period and terminal repolarization period are summarized in Fig. 3, of which pre-drug basal control values (C) were 247 ± 4, 227 ± 9, 204 ± 6 and 43 ± 9 ms, respectively. The low or middle dose did not alter any of the variables. The high dose prolonged the effective refractory period for 45–60 min, whereas no significant change was detected in the other variables.
Discussion
We performed a reverse translational research to analyze the onset mechanisms of cardiovascular adverse events of lamotrogine by assessing its electropharmacological effects on the in situ hearts, and obtained several interesting findings as discussed below.
Electropharmacological effects
Lamotrigine in any dose examined did not alter the hemodynamic variables, indicating that lamotrigine has relatively wide safety margin for them, and that clinically reported hypotension may not be induced through its direct action on the resistance arterioles or capacitance venules [1]. Meanwhile, the high dose of lamotrigine significantly delayed the intra-atrial and intra-ventricular conductions in addition to the prolongation of ventricular effective refractory period, suggesting that lamotrigine can inhibit Na+ channel in the in situ hearts. However, such in vitro effects on cardiac ionic channels have not been studied as far as we know. Thus, these in vivo electrophysiological findings may partly explain the onset mechanism of lamotrigine-associated elevation of the atrial pacing threshold, cardiac conduction delay, wide complex tachycardia and cardiac arrest in clinical cases [1,2,3].
We found that lamotrigine did not delay the ventricular repolarization, which would bridge the gap between the “clinical experience” that lamotrigine has not induced torsade de pointes and the “basic research” that lamotrigine inhibited IKr with large IC50 value of 229 µM in human embryo kidney 293 cells [10] which would be > 30 times greater than its unbound free therapeutic plasma concentration. On the other hand, lamotrigine has been clinically shown to induce Brugada-like electrocardiographic pattern in 3 cases [3,4,5] and J-wave-like electrocardiographic abnormality in 2 cases [17]. Since lamotrigine was shown to be able to inhibit the ventricular Na+ channels in vivo, and to elevate J wave in half of the animals studied, lamotrigine may have potential to unmask Brugada electrocardiographic genotype in susceptible patients.
Study limitation
First, higher dose of more than 10 mg/kg would be required to clarify the pharmacological effect of lamotrigine, since estimated Cmax in this study after the administration of 10 mg/kg could be approximately 3 times greater than that after single oral dose of 200 mg in human subjects, and more higher plasma concentrations might be expected in patients with impaired hepatic and/or renal function. Second, we did not evaluate the rate-dependent effect of lamotrigine on QRS width, which may further confirm that lamotrigine can directly block the voltage-gated Na+ channel. Third, lamotrigine inhibited Na+ channels in the in situ hearts, which may support the depolarization hypothesis of J wave appearance. However, since we did not simultaneously record the local action potentials at the epicardial and endocardial surfaces, the currently obtained results may not provide enough information to support the repolarization hypothesis, but might at least support the utility of J wave appearance as a marker for predicting the drug-induced early repolarization in susceptible patients.
Conclusion
While lamotrigine may have wide safety margin for hemodynamic variables, its supratherapeutic concentration will inhibit Na+ channel in the in situ hearts, explaining clinically reported lamotrigine-associated cardiac adverse events. It should be also noted that lamotrigine by itself will not be associated with the onset of torsade de pointes, but might induce Brugada syndrome in some patients having genetic risk factors.
References
Alyahya B, Friesen M, Nauche B, Laliberté M (2018) Acute lamotrigine overdose: a systematic review of published adult and pediatric cases. Clin Toxicol 56:81–89
Irifune R, Matsuda M, Tagaya M, Hara K, Tamura R (2017) Marked elevation of the atrial pacing threshold in a patient with sinus dysfunction on lamotrigine therapy. Open J Clin Med Case Rep 3:1299
Strimel WJ, Woodruff A, Cheung P, Kirmani BF, Stephen Huang SK (2010) Brugada-like electrocardiographic pattern induced by lamotrigine toxicity. Clin Neuropharmacol 33:265–267
Rodrigues R, Amador P, Rassi L, Seixo F, Parreira L, Fonseca N, Soares L (2013) Brugada pattern in a patient medicated with lamotrigine (article in Portuguese; title and abstract were also provided in English). Rev Port Cardiol 32:807–810
Leong KMW, Seligman H, Varnava AM (2017) Proarrhythmogenic effects of lamotrigine during ajmaline testing for Brugada syndrome. HeartRhythm Case Rep 3:167–171
Lang DG, Wang CM, Cooper BR (1993) Lamotrigine, phenytoin and carbamazepine interactions on the sodium current present in N4TG1 mouse neuroblastoma cells. J Pharmacol Exp Ther 266:829–835
Xie X, Lancaster B, Peakman T, Garthwaite J (1995) Interaction of the antiepileptic drug lamotrigine with recombinant rat brain type IIA Na+ channels and with native Na+ channels in rat hippocampal neurons. Pflügers Arch 430:437–446
Zona C, Avoli M (1997) Lamotrigine reduces voltage-gated sodium currents in rat central neurons in culture. Epilepsia 38:522–525
Hainsworth AH, McNaughton NCL, Pereverzev A, Schneider T, Randall A (2003) Actions of sipatrigine, 202W92 and lamotrigine on R-type and T-type Ca2+ channel currents. Eur J Pharmacol 467:77–80
Polak S, Wisniowska B, Brandys J (2009) Collation, assessment and analysis of literature in vitro data on hERG receptor blocking potency for subsequent modeling of drugs’ cardiotoxic properties. J Appl Toxicol 29:183–206
Sugiyama A (2008) Sensitive and reliable proarrhythmia in vivo animal models for predicting drug-induced torsades de pointes in patients with remodelled hearts. Br J Pharmacol 154:1528–1537
Haïssaguerre M, Derval N, Sacher F, Jesel L, Deisenhofer I, de Roy L, Pasquié JL, Nogami A, Babuty D, Yli-Mayry S, De Chillou C, Scanu P, Mabo P, Matsuo S, Probst V, Le Scouarnec S, Defaye P, Schlaepfer J, Rostock T, Lacroix D, Lamaison D, Lavergne T, Aizawa Y, Englund A, Anselme F, O’Neill M, Hocini M, Lim KT, Knecht S, Veenhuyzen GD, Bordachar P, Chauvin M, Jais P, Coureau G, Chene G, Klein GJ, Clémenty J (2008) Sudden cardiac arrest associated with early repolarization. N Engl J Med 358:2016–2023
Yamazaki-Hashimoto Y, Nakamura Y, Ohara H, Cao X, Kitahara K, Izumi-Nakaseko H, Ando K, Yamazaki H, Ikeda T, Yamazaki J, Sugiyama A (2015) Fluvoxamine by itself has potential to directly induce long QT syndrome at supra-therapeutic concentrations. J Toxicol Sci 40:33–42
Van de Water A, Verheyen J, Xhonneux R, Reneman RS (1989) An improved method to correct the QT interval of the electrocardiogram for changes in heart rate. J Pharmacol Methods 22:207–217
Sugiyama A, Hashimoto K (1998) Effects of gastrointestinal prokinetic agents, TKS159 and cisapride, on the in situ canine heart assessed by cardiohemodynamic and electrophysiological monitoring. Toxicol Appl Pharmacol 152:261–269
Takahara A, Wagatsuma H, Aritomi S, Konda T, Akie Y, Nakamura Y, Sugiyama A (2011) Measurements of cardiac ion channel subunits in the chronic atrioventricular block dog. J Pharmacol Sci 116:132–135
Ishizue N, Niwano S, Saito M, Fukaya H, Nakamura H, Igarashi T, Fujiishi T, Yoshizawa T, Oikawa J, Satoh A, Kishihara J, Murakami M, Niwano H, Miyaoka H, Ako J (2016) Polytherapy with sodium channel-blocking antiepileptic drugs is associated with arrhythmogenic ST-T abnormality in patients with epilepsy. Seizure 40:81–87
Acknowledgements
This study was supported in part by JSPS KAKENHI (Grant Numbers 19K16505 and 20K16136). The authors thank Mrs. Yuri Ichikawa for her technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors indicated no potential conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Goto, A., Hagiwara-Nagasawa, M., Kambayashi, R. et al. Reverse translational analysis of clinically reported, lamotrigine-induced cardiovascular adverse events using the halothane-anesthetized dogs. Heart Vessels 36, 424–429 (2021). https://doi.org/10.1007/s00380-020-01716-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00380-020-01716-8