
Abstract
Lithium is one of the classical drugs that have been widely used for treating bipolar disorder. However, several cardiac side effects including sick sinus syndrome, bundle branch block, ventricular tachycardia/fibrillation, non-specific T-wave abnormalities in addition to Brugada-type electrocardiographic changes have been noticed in patients who were given antidepressant, anticonvulsant, and/or antipsychotic drugs besides lithium. In this study, we assessed cardiohemodynamic and electrophysiological effects of lithium carbonate by itself to begin to analyze onset mechanisms of its cardiovascular side effects. Lithium carbonate in intravenous doses of 0.1, 1, and 10 mg/kg over 10 min was cumulatively administered with an interval of 20 min to the halothane-anesthetized beagle dogs (n = 4), which provided peak plasma Li+ concentrations of 0.02, 0.18, and 1.79 mEq/L, respectively, reflecting sub-therapeutic to toxic concentrations. The low and middle doses prolonged the ventricular effective refractory period at 30 min and for 5–30 min, respectively. The high dose decreased the heart rate for 45–60 min, delayed the intraventricular conduction for 15–20 min and the ventricular repolarization at 45 min, and prolonged the effective refractory period for 5–60 min. No significant change was detected in the other cardiovascular variables. Thus, lithium alone may have a wide safety margin against hemodynamic adverse events; however, it would directly and/or indirectly inhibit Na+ and K+ channels, which may synergistically increase the ventricular refractoriness from the sub-therapeutic concentration and decrease the heart rate at the supra-therapeutic one. These findings may partly explain its clinically observed various types of arrhythmias as well as electrocardiographic changes.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Lithium is one of the classical drugs that have been widely used for the treatment of bipolar disorder. However, several cardiac side effects have been reported including the bradycardia-tachycardia (“sick” sinus) syndrome, atrial flutter, atrioventricular block, bundle branch block, ventricular tachycardia/fibrillation in addition to non-specific T-wave abnormalities in electrocardiogram [1, 2]. More importantly, Brugada-type electrocardiographic changes have been reported in three patients with long-term treatment of lithium [3, 4]. Two out of three patients showed serum Li+ level of 1.0 and 2.5 mEq/L (therapeutic range 0.8–1.2), respectively, who were also given antidepressant, anticonvulsant and/or antipsychotic drugs in combination with lithium, whereas no information is available for the last patient [3, 4]. Since most of the cardiac side effects of lithium are noticed in patients who were also treated with other drugs, effects of lithium by itself on the cardiovascular system need to be clarified to begin to analyze the onset mechanisms of its cardiovascular side effects, which have not been fully investigated before.
In this study, we simultaneously assessed the cardiohemodynamic and electrophysiological effects of lithium carbonate using the halothane-anesthetized beagle dogs, which have been known to be able to predict the electropharmacological responses in healthy human subjects [5]. In order to better analyze the proarrhythmic potential of lithium carbonate, we analyzed early (J–Tpeak) and late (Tpeak–Tend) repolarization periods in the electrocardiogram. The duration of early repolarization has been considered to reflect the net balance between the inward Na+ and Ca2+ currents and the outward K+ current [6, 7], of which prolongation may indicate intracellular Ca2+ overload. Meanwhile, the prolongation of late repolarization can estimate the extent of inhibition for human ether-à-go-go-related gene (hERG) K+ channel [6, 7]. Furthermore, we simultaneously measured monophasic action potential (MAP) and effective refractory period at the same site to predict the terminal repolarization period, which can quantify the risk of a drug to induce reentrant ventricular arrhythmias [5].
Materials and Methods
Experiments were performed in female beagle dogs weighing approximately 10 kg (n = 4). Animals were obtained through Kitayama Labes Co., Ltd. (Nagano, Japan). The dogs were initially anesthetized with thiopental sodium (30 mg/kg, i.v.). After intubation with a cuffed endotracheal tube, 1% halothane vaporized with 100% oxygen was inhaled with a volume-limited ventilator (SN-480-3; Shinano Manufacturing Co., Ltd., Tokyo, Japan). Tidal volume and respiratory rate were set at 20 mL/kg and 15 breaths/min, respectively.
Cardiohemodynamic Variables
Four clinically available catheter-sheath sets (FAST-CATH™ 406119; St. Jude Medical Daig Division, Inc., Minnetonka, MN, USA) were used; two were inserted into the right and left femoral arteries toward abdominal aorta; the other two were done into the right and left femoral veins toward inferior vena cava, respectively. A pig-tail catheter was placed at the left ventricle to measure the left ventricular pressure through the right femoral artery, whereas blood pressure was measured at a space between inside of the catheter sheath and outside of the pig-tail catheter through a flush line. The maximum upstroke velocities of the left ventricular pressure and the left ventricular end-diastolic pressure were obtained during sinus rhythm to estimate the inotropic status and the preload to left ventricle, respectively. A thermodilution catheter (132F5; Edwards Lifesciences, Irvine, CA, USA) was positioned at the right side of the heart through the right femoral vein. The cardiac output was measured with a standard thermodilution method by using a cardiac output computer (MFC-1100; Nihon Kohden Co., Ltd., Tokyo, Japan). The total peripheral vascular resistance was calculated with the basic equation: total peripheral vascular resistance = mean blood pressure/cardiac output.
Electrophysiological Variables
The surface lead II electrocardiogram was obtained from the limb electrodes. Corrected QT interval (QTc) was calculated with Van de Water’s formula: QTc = QT − 0.087 × (RR − 1000) with RR given in ms [8]. A standard six French quad-polar electrodes catheter (Cordis-Webster Inc., Baldwin Park, CA, USA) was positioned at the non-coronary cusp of the aortic valve through the left femoral artery to obtain the His bundle electrogram.
A bi-directional steerable MAP recording/pacing combination catheter (1675P; EP Technologies, Inc., Sunnyvale, CA, USA) was positioned at the endocardium of the right ventricle through the left femoral vein to obtain MAP signals. The signals were amplified with a DC preamplifier (model 300; EP Technologies, Inc.). The interval (ms) at 90% repolarization level was defined as MAP90. The heart was electrically driven by using a cardiac stimulator (SEC-3102; Nihon Kohden Co., Ltd.) through the pacing electrodes of the combination catheter placed in the right ventricle. The stimulation pulses were rectangular in shape 1–2 V of amplitude (about twice the threshold voltage) and of 1-ms duration. The MAP90 was measured during sinus rhythm [MAP90(sinus)] and at a pacing cycle length of 400 ms [MAP90(CL400)] and 300 ms [MAP90(CL300)].
The effective refractory period of the right ventricle was assessed with programmed electrical stimulation. The pacing protocol consisted of five beats of basal stimuli in a cycle length of 400 ms followed by an extra stimulus of various coupling intervals. The duration of the terminal repolarization period of the ventricle was calculated by the difference between the MAP90(CL400) and effective refractory period at the same site, which reflects the extent of electrical vulnerability of the ventricular muscle [5].
The J–Tpeak and Tpeak–Tend were measured separately [7]. When the end of T-wave was obscure, we used MAP signal as a guide to estimate it. The J–Tpeak was corrected for the heart rate with a coefficient as previously described (J–Tpeakc = J–Tpeak/RR0.58 with RR given in seconds) [6]. Correction was not performed on the Tpeak–Tend, since previous QT/QTc studies have shown that the Tpeak–Tend exhibited minimal heart rate dependency at the resting heart rate [6].
Experimental Protocol
The aortic and left ventricular pressures, electrocardiogram, His-bundle electrogram, and MAP signals were monitored with a polygraph system (RM-6000; Nihon Kohden Co., Ltd.) and analyzed with a real-time fully automatic data analysis system (WinVAS3 ver. 1.1R24; Physio-Tech Co., Ltd., Tokyo, Japan). Each measurement of electrocardiogram and MAP variables as well as atrio-His (AH) and His-ventricular (HV) intervals adopted the mean of three recordings of consecutive complexes. After the basal assessment, lithium carbonate in a low dose of 0.1 mg/kg was intravenously administered over 10 min, and each variable was assessed at 5, 10, 15, 20, and 30 min after the start of infusion. Then, lithium carbonate in a middle dose of 1 mg/kg was intravenously administered over 10 min, and each variable was assessed in the same manner. Finally, lithium carbonate in a high dose of 10 mg/kg was intravenously administered over 10 min, and each variable was assessed at 5, 10, 15, 20, 30, 45, and 60 min after the start of infusion.
Plasma Drug Concentration
Blood was sampled from the left femoral artery at 5, 10, 15, and 30 min after the start of the low and middle doses infusion, and 5, 10, 15, 30, and 60 min after the start of the high-dose infusion. The blood samples were centrifuged at 1500×g for 15 min at 4 °C. The plasma was stored at − 80 °C until the drug concentration was measured. The plasma drug concentration was detected as Li+, which was measured by atomic absorption spectrometry at LSI Medience Corporation (Tokyo, Japan). The detection limit of Li+ was 0.01 mEq/L.
Drugs
Lithium carbonate (Sigma-Aldrich Co., LLC., St. Louis, MO, USA) was dissolved with saline in concentrations of 0.1, 1, and 10 mg/mL. Other drugs used were thiopental sodium (Ravonal® 0.5 g for Injection, Mitsubishi Tanabe Pharma Co., Osaka, Japan), halothane (Fluothane®, Takeda Pharmaceutical Co., Ltd., Osaka, Japan), and heparin calcium (HEPARIN Ca, Sawai Pharmaceutical Co., Ltd., Osaka, Japan).
Statistical Analysis
Data are presented as mean ± SEM. Differences within a parameter were evaluated with one-way, repeated-measures analysis of variance (ANOVA) followed by Contrasts as a post hoc-test for mean values comparison. A p value < 0.05 was considered to be significant.
Results
No animals exerted any lethal ventricular arrhythmias or hemodynamic collapse leading to the animals’ death during the experiment.
Effects on the Cardiohemodynamic Variables
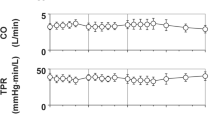
Typical tracings of the aortic and left ventricular pressures are depicted in Fig. 1, and the time courses of changes in the plasma concentration of Li+ and the cardiohemodynamic variables are summarized in Fig. 2. The peak plasma concentrations of Li+ were observed at 10 min after the start of 0.1, 1 and 10 mg/kg infusion, which were 0.02 ± 0.00, 0.18 ± 0.02, and 1.79 ± 0.17 mEq/L, respectively. The pre-drug basal control values (C) of the heart rate, mean blood pressure, cardiac output, total peripheral vascular resistance, maximum upstroke velocity of left ventricular pressure, and left ventricular end-diastolic pressure were 103 ± 10 beats/min, 105 ± 6 mmHg, 2.4 ± 0.2 L/min, 46 ± 5 mmHg min/L, 1,887 ± 267 mmHg/s, and 10 ± 2 mmHg, respectively. The low and middle doses did not alter any of the variables. The high dose decreased the heart rate for 45–60 min after the start of infusion, whereas no significant change was detected in the other variables.

Typical tracings showing the His-bundle electrogram (His), lead II electrocardiogram (ECG), aortic pressure (AoP), left ventricular pressure (LVP), and monophasic action potential (MAP) during sinus rhythm at pre-drug basal control (control, left) and 45 min after the start of 10 mg/kg of lithium carbonate infusion (45 min after 10 mg/kg i.v., right)

Time courses of changes in the plasma concentration of Li+, heart rate (HR), mean blood pressure (MBP), cardiac output (CO), total peripheral vascular resistance (TPR), maximum upstroke velocity of the left ventricular pressure (LVdP/dtmax), and left ventricular end-diastolic pressure (LVEDP) after the administration of lithium carbonate (n = 4). Data are presented as mean ± SEM. Closed symbols represent significant differences from the corresponding pre-drug basal control value (C) by p < 0.05. Since we could not obtain actual values of Li+ at 5, 15, and 30 min after the start of administration of 0.1 mg/kg of lithium carbonate possibly due to the lower concentrations than its detection limit of 0.01 mEq/L, we plotted these results as 0.01 mEq/L using gray symbol line
Effects on the Electrocardiographic Variables
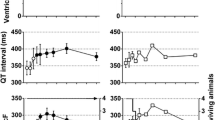
Typical tracings of the electrocardiogram are depicted in Fig. 1, and the time courses of changes in the electrocardiographic variables are summarized in Fig. 3. The pre-drug basal control values (C) of the PR interval, QRS width, QT interval, QTc, J–Tpeakc, and Tpeak–Tend were 111 ± 5, 66 ± 2, 266 ± 19, 301 ± 14, 158 ± 10, and 84 ± 17 ms, respectively. The low, middle, or high dose did not alter any of the variables.

Time courses of changes in the PR interval, QRS width, QT interval, QTc, J–Tpeakc, Tpeak–Tend; the atrio-His (AH) and His-ventricular (HV) intervals, and MAP90 during sinus rhythm [MAP90(sinus)]; and the MAP90 at a cycle length of 400 ms [MAP90(CL400)] and 300 ms [MAP90(CL300)], effective refractory period of the right ventricle (ERP) and terminal repolarization period (TRP) after the administration of lithium carbonate (n = 4). QT interval was corrected with Van de Water’s formula: QTc = QT − 0.087 × (RR − 1000). J–Tpeak was corrected by the following formula: J–Tpeakc = J–Tpeak/RR0.58. MAP90: monophasic action potential duration at 90% repolarization level. Data are presented as mean ± SEM. Closed symbols represent significant differences from the corresponding pre-drug basal control value (C) by p < 0.05
Effects on the AH and HV Intervals, and MAP90 During Sinus Rhythm
Typical tracings of the His-bundle electrogram and MAP are depicted in Fig. 1, and the time courses of changes in the AH and HV intervals, and MAP90 during sinus rhythm are summarized in Fig. 3. Their pre-drug basal control values (C) were 80 ± 7, 27 ± 2, and 253 ± 17 ms, respectively. The low or middle dose did not alter any of the variables. The high dose prolonged the HV interval for 15–20 min, whereas no significant change was detected in the other variables.
Effects on the MAP90 During the Ventricular Pacing, Effective Refractory Period, and Terminal Repolarization Period
Typical tracings showing the aortic and left ventricular pressures and MAP during the ventricular programmed electrical stimulation at a basic cycle length of 400 ms in Fig. 4. The time courses of changes in the MAP90(CL400), MAP90(CL300), effective refractory period and terminal repolarization period are summarized in Fig. 3, of which pre-drug basal control values (C) were 245 ± 14, 229 ± 11, 209 ± 10, and 36 ± 4 ms, respectively. The low and middle doses prolonged the effective refractory period at 30 min and for 5–30 min, respectively, whereas no significant change was detected in the other variables. The high dose prolonged the MAP90(CL400), MAP90(CL300), and effective refractory period at 45 min, at 45 min, and for 5–60 min, respectively, whereas no significant change was detected in the terminal repolarization period.

Typical tracings showing the aortic (AoP) and left ventricular (LVP) pressures and monophasic action potentials (MAP) during the ventricular programmed electrical stimulation at a basic cycle length (CL) of 400 ms, which estimate the ventricular effective refractory period (ERP) at pre-drug control (Control) and 45 min after the start of 10 mg/kg, i.v. of lithium carbonate infusion (45 min after 10 mg/kg i.v.). At pre-drug control (upper panels), premature electrical stimulation (pacing) with a coupling interval (CI) of 190 ms induced ventricular electrical activity (left), whereas that of 185 ms failed to develop it (right), indicating that the ERP was 190 ms. After the administration of lithium carbonate (lower panels), premature electrical stimulation with a CI of 205 ms induced ventricular electrical activity (left), whereas that of 200 ms did not develop it (right), indicating that the ERP was 205 ms
Discussion
In this study, we analyzed the onset mechanisms of the cardiovascular side effects of lithium carbonate by assessing its cardiovascular profile, and obtained several interesting findings as discussed below.
Rationale of the Drug Dose
According to the interview form published September 2012, version 5 from the manufacturer (Taisho Pharmaceutical Co. Ltd., Tokyo, Japan), initial oral dose of lithium carbonate is 400–600 mg/day in 2–3 divided doses, which can be gradually elevated to 1200 mg/day, and the Cmax of Li+ after a single oral administration of 200 mg/body was 0.22 mEq/L. Meanwhile, the target plasma concentration has been clinically considered to be 0.6–1.4 mEq/L [1]. Since the peak plasma concentrations of Li+ after the administration of 0.1, 1, and 10 mg/kg were 0.02, 0.18, and 1.79 mEq/L, respectively, in this study, currently used doses would provide sub-therapeutic to toxic plasma concentrations.
Cardiohemodynamic Effects
The low to high doses of lithium carbonate did not alter the cardiohemodynamic variables except that the high dose decreased the heart rate for 45–60 min after its administration. These observations may partly support a previous finding by an isolated, perfused heart preparation that Li+ has little acute, direct effect on the function of the heart [9]. Since Li+ is closely related to Na+ in its properties, it could substitute for Na+ both in generating action potentials and in Na+-Na+ exchange across the membrane, whereas it does not significantly affect the Na+-Ca2+ exchanger or the Na+/K+-ATPase pump at therapeutic concentrations [1]. Importantly, as discussed below, Li+ may inhibit both Na+ and K+ channels, which may explain the late onset of bradycardic action of this study, that might be partly related to clinically reported sick sinus syndrome. Nonetheless, these results at least suggest that lithium carbonate may have a certain extent of safety margin against the hemodynamic adverse events except for the bradycardic action.
Electrophysiological Effects
Lithium carbonate did not alter the atrioventricular nodal conduction at any dose, but its high dose prolonged the intraventricular conduction time and repolarization period. Importantly, lithium carbonate prolonged the effective refractory period from the low-dose administration. These results suggest that lithium carbonate may moderately inhibit both Na+ and K+ channels, which might have prolonged the effective refractory period through a synergetic mechanism. Qualitatively similar action of lithium gluconate on the atrial effective refractory period has been reported [10]. In a previous study using Chinese hamster ovary cells transfected with SCN5A encoding cardiac Na+ channel, lithium chloride suppressed INa with IC50 values of 6.8 µmol/L [3]. Since lithium does not bind serum protein [1], currently obtained in vivo results did not reflect such a potent in vitro INa inhibition of lithium. Further study is needed to clarify the difference in the potency of INa inhibition between in vivo and in vitro experiments, which can be approximately 1000 times less potent in the former than the latter. In addition, another potential mechanism may include the lithium-induced increase of plasma K+ level [11], although we did not measure it in this study. Hyperkalemia can depolarize the resting membrane potential through a change in the K+ equilibrium potential. If the depolarization is great enough, it may inactivate Na+ channels, resulting in increased refractoriness and slowed ventricular conduction.
Proarrhythmic Effects
The high dose of lithium carbonate delayed the repolarization period in this study, which is qualitatively in accordance with a clinical study that Li+ concentration correlates with QTc in patients with psychosis [12]. However, lithium carbonate did not alter the J–Tpeakc, Tpeak–Tend or terminal repolarization period. These results indicate that lithium carbonate by itself may not possess electrophysiological properties leading to the onset of long QT-interval related, lethal ventricular arrhythmias [6, 7], which may explain why there are no reported case of lithium-induced torsade de pointes [13].
Conclusion
Lithium carbonate by itself may have a wide safety margin against hemodynamic adverse events; however, it would directly and/or indirectly inhibit both Na+ and K+ channels, leading to the increase of ventricular refractoriness from the sub-therapeutic concentration and the decrease of heart rate at the supra-therapeutic one. These findings may partly explain its clinically observed adverse events of various types of arrhythmias as well as electrocardiographic changes. In order to further analyze the onset mechanisms of adverse effects of lithium, cross-reaction between lithium and other medications, background pathophysiology of patients who had adverse events, and long-term effects should be clarified.
References
DeBattista, C. (2015). Antipsychotic agents & lithium. In B. G. Katzung & A. J. Trevor (Eds.), Basic & clinical pharmacology (13th ed., pp. 490–509). New York: McGraw Hill Education.
Yap, Y. G., Behr, E. R., & Camm, A. J. (2009). Drug-induced Brugada syndrome. Europace, 11, 989–994.
Darbar, D., Yang, T., Churchwell, K., Wilde, A. A., & Roden, D. M. (2005). Unmasking of brugada syndrome by lithium. Circulation, 112, 1527–1531.
Wright, D., & Salehian, O. (2010). Brugada-type electrocardiographic changes induced by long-term lithium use. Circulation, 122, e418–e419.
Sugiyama, A. (2008). Sensitive and reliable proarrhythmia in vivo animal models for predicting drug-induced torsades de pointes in patients with remodelled hearts. British Journal of Pharmacology, 154, 1528–1537.
Johannesen, L., Vicente, J., Mason, J. W., Sanabria, C., Waite-Labott, K., Hong, M., et al. (2014). Differentiating drug-induced multichannel block on the electrocardiogram: randomized study of dofetilide, quinidine, ranolazine, and verapamil. Clinical Pharmacology & Therapeutics, 96, 549–558.
Cao, X., Nakamura, Y., Wada, T., Izumi-Nakaseko, H., Ando, K., & Sugiyama, A. (2016). Electropharmacological effects of amantadine on cardiovascular system assessed with J-Tpeak and Tpeak-Tend analysis in the halothane-anesthetized beagle dogs. The Journal of Toxicological Sciences, 41, 439–447.
Van de Water, A., Verheyen, J., Xhonneux, R., & Reneman, R. S. (1989). An improved method to correct the QT interval of the electrocardiogram for changes in heart rate. Journal of Pharmacological Methods, 22, 207–217.
Linakis, J. G., Savitt, D. L., Schuyler, J. E., Simon, P. M., & Raymond, R. M. (2000). Lithium has no direct effect on cardiac function in the isolated, perfused rat heart. Pharmacology & Toxicology, 87, 39–45.
Descotes, J., Lievre, M., Rouzioux, J. M., Faucon, G., & Evreux, J. C. (1978). Cardiac electrophysiological effects of lithium gluconate in anesthetized dogs. Therapie, 33, 661–670; (in French).
Abdel-Zaher, A. O., & Abdel-Rahman, M. M. (1999). Lithium chloride-induced cardiovascular changes in rabbits are mediated by adenosine triphosphate-sensitive potassium channels. Pharmacological Research, 39, 275–282.
Mamiya, K., Sadanaga, T., Sekita, A., Nabeyama, Y., Yao, H., & Yukawa, E. (2005). Lithium concentration correlates with QTc in patients with psychosis. Journal of Electrocardiology, 38, 148–151.
Alvarez, P. A., & Pahissa, J. (2010). QT alterations in psychopharmacology: proven candidates and suspects. Current Drug Safety, 5, 97–104.
Acknowledgements
The authors thank Ms. Misako Nakatani and Mrs. Yuri Ichikawa for their assistance during preparation of the manuscript.
Funding
This study was supported in part by JSPS KAKENHI Grant No. JP16K08559 and AMED Grant No. JP17am0101122.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors indicated no potential conflict of interest.
Ethical Approval
All experiments were approved by the Toho University Animal Care and User Committee (No. 17-54-275) and performed in accordance with the Guidelines for the Care and Use of Laboratory Animals of Toho University.
Additional information
Handling Editor: Kurt J. Varner.
Rights and permissions
About this article

Cite this article
Goto, A., Nakamura, Y., Lubna, N.J. et al. Analysis of Safety Margin of Lithium Carbonate Against Cardiovascular Adverse Events Assessed in the Halothane-Anesthetized Dogs. Cardiovasc Toxicol 18, 530–536 (2018). https://doi.org/10.1007/s12012-018-9464-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12012-018-9464-0