
Abstract
Hypoxia/reoxygenation (H/R) is one of the cellular stresses in pathological conditions, such as myocardial infarction, stroke and organ transplantation. Oxidative stress caused by reactive oxygen species (ROS) is a crucial element of H/R injury in vascular endothelial cells (ECs). Xanthine oxidase (XO) has been recognized to contribute to H/R injury. Of note, xanthine oxidoreductase is synthesized as xanthine dehydrogenase (XDH) and needs to be converted to XO to become a source of superoxide. Hepatocyte growth factor (HGF) has been found to protect ECs against H/R injury. The relation, however, between HGF and XO in ECs under H/R conditions remains to be determined. Primary cultured rat cardiac microvascular endothelial cells (CMECs) were exposed to 4 h of hypoxia and followed by 1 h of reoxygenation. Generation of ROS and cytosolic Ca2+ concentration was measured by flow cytometry qualification of DCFHDA and fluo-3 AM staining cells, respectively. XDH mRNA was qualified by qRT-PCR analysis. XO activity was determined by colorimetric assay and XO protein levels were determined by Western blot. Cell apoptosis was assessed by caspase-3 activity and Annexin V/PI staining. After H/R, cellular ROS production significantly increased. Both XO activity and XO protein increased after H/R. Cellular ROS elevation was inhibited by allopurinol (a potent XO inhibitor), indicting XO accounting for the generation of ROS after H/R. In addition, XDH mRNA increased after H/R, indicating a de novo XDH synthesis, which needs to be converted to XO to become a source of superoxide. Pretreatment of HGF inhibited the elevation of XO activity and XO protein level after H/R; however, HGF has no effect on the increase of XDH mRNA. We also find an increase of the cytosolic Ca2+ in CMECs after H/R. BAPTA-AM, a cell-permeable Ca2+ chelator, prevented the increase of XO activity and XO protein levels, implicating the elevated cytosolic Ca2+ concentration involvement in XO conversion and XO activation. HGF inhibited the elevation of cytosolic Ca2+ concentration in CMECs after H/R. Furthermore, HGF ameliorated H/R-induced CMECs apoptosis. These findings suggest a novel mechanism whereby HGF inhibited XO-generated ROS production after H/R treatment. H/R induces a de novo synthesis of XDH, the XO precursor. In addition, H/R increases cytosolic Ca2+ concentration and promotes a Ca2+-involved XO conversion and XO activation. HGF has no effect on the increase of XDH mRNA; however, HGF inhibited the elevation of XO protein level and XO activity after H/R in the post-transcriptional level primarily by inhibiting the increase of cytosolic Ca2+ concentration. HGF protects CMECs from H/R-induced apoptosis by inhibiting the elevation of XO protein level and XO activity.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Hypoxia/reoxygenation (H/R) is one of the cellular stresses in pathological conditions, such as myocardial infarction, stroke and organ transplantation. Oxidative stress caused by reactive oxygen species (ROS) is a crucial element of pathophysiological process of H/R injury [1]. Xanthine oxidase (XO) has been widely recognized to contribute to H/R injury by generating superoxide anions (O2 −) in various cell types [2, 3]. Xanthine oxidoreductase (XOR) is a generic term for two distinct enzyme forms: xanthine dehydrogenase (XDH) and XO. XDH is constitutively expressed and the post-transcriptionally modified form XO is seen at high concentrations under H/R conditions [4]. XDH requires NAD+ as an electron acceptor to reduce hypoxanthine to xanthine, thereby generating the stable reaction product NADH. XO requires the reduction of O2 for purine oxidation, thereby generating the highly reactive superoxide free radical (O2 −) [5]. XOR is found to be localized on the luminal surface of the microvascular endothelium of many organs such as heart, and XO has been identified as a major endothelial source of ROS that is activated in atherosclerosis, coronary disease and heart failure [6].
The multifunctional protein hepatocyte growth factor (HGF), also named scatter factor, exerts potent proliferation, survival, motility and morphogenesis activities on various cell types by activating the receptor tyrosine kinase cMet. The binding of HGF to cMet has been found to protect against H/R injury in murine lung endothelial cells [7], rat cerebral endothelial cells [8], and in vitro mouse lung [9]. HGF ameliorated endothelial dysfunction and mediated enhanced proliferation of endothelial cells in rat lungs [10]. In addition, HGF suppresses oxidative stress and ROS production in high glucose-treated rat mesangial cells [11] and in advanced glycation end product-treated human umbilical vein endothelial cells [12].
However, whether HGF affects ROS production and oxidative stress under H/R conditions remains to be determined. Given that H/R leads to changes in O2 levels, we hypothesized that ROS generated by XO contributes to H/R injury in rat cardiac microvascular endothelial cells (CMECs). The aim of this study was to evaluate the effect of HGF on XO activity in CMECs under H/R conditions and to elucidate the possible mechanism of this effect.
Materials and methods
Cardiac microvascular endothelial cells isolation and culture
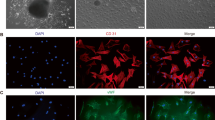
Sprague–Dawley (SD) rats aged 5–7 days, weight 12–16 g, were purchased from the Experimental Animal Center of PLA General Hospital, Beijing, China (approval No. SCXK 20120001). 130 SD rats were sacrificed in this study. All procedures were conducted in conformity with the National Institutes of Health Guideline on the Use of Laboratory Animals and all experiments were performed in accordance with the Helsinki declaration. CMECs were isolated from neonatal SD rat hearts by enzyme dissociation method based on the one described by Nishida [13]. Briefly, neonatal rats were euthanized using isoflurane. Hearts were removed under sterile conditions. After removing atria, visible connective tissue, right ventricle and valvular tissue, left ventricle was immersed in 75 % ethanol for 20 s to devitalize epicardial and endocardial endothelial cells. The outer one-fourth ventricular wall was peeled away. The remaining tissue was minced into pieces of 0.5 mm × 0.5 mm × 0.5 mm and digested in 0.5 % (w/v) collagenase type I (Gibco, #17100-017) for 20 min and 0.125 % (w/v) trypsin (Hyclone, #SH30042.01) for 10 min at 37 °C in a shaking bath, the solution was filtered through an 100-μM nylon mesh to remove undigested tissue. The dissociated cells were resuspended in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco, #12100046) supplemented with 20 % (v/v) fetal bovine serum (FBS) (Hyclone, #YS-SO-001091) and then seeded in 25 cm2 polystyrene flasks (Nunc, #156340). Cultured cell purity was tested by their morphological and immunohistochemistry characteristics [14]: the CMECs monolayer displayed uniform ‘cobblestone’ morphology and immunohistochemistry assay of factor VIII (Abcam, #ab61910) and CD31 (XINXINGTANG BIOTECHNOLOGY, #Xt-0468R) demonstrated positive (>95 %).
Hypoxia/reoxygenation procedure and drug treatment
Hypoxic conditions were produced using D-Hanks solution (in mM: 5.37 KCl, 0.44 KH2PO4, 136.89 NaCl, 4.166 NaHCO3, 0.338 Na2HPO4, pH 7.3–7.4 at 37 °C) saturated with 95 % N2 and 5 % CO2. The pH was adjusted to 6.8 with lactate to mimic ischemic conditions. The dishes were put into a hypoxic incubator (Invivo2-400, Ruskinn) that was equilibrated with 95 % N2 and 5 % CO2. Ambient O2 levels in the hypoxia incubator were monitored by an O2 analyzer (series-2000, Alpha Omega Instruments). After hypoxic treatment, the culture medium was rapidly replaced with fresh DMEM with 1 % FBS to initiate reoxygenation [15]. Hypoxia/reoxygenation procedure was achieved by 4 h of hypoxia treatment and 1 h of reoxygenation treatment.
For drug treatment, cultured CMECs were pre-incubated with 10 ng/ml or 20 ng/ml HGF (RnD, #294-HG-005) for 30 min before hypoxia. To determine the effect of XO on CMECs endogenous ROS production, XO inhibitor allopurinol was diluted in D-Hanks solution at a final concentration of 20 or 40 μM and pretreated CMECs for 30 min before hypoxia. To determine the effect of cytosolic Ca2+ on endogenous XO production and activation, a cell-permeable Ca2+ chelator BAPTA-AM (Sigma, #A1076) was diluted in D-Hanks solution at a final concentration of 1 μM and pretreated CMECs for 30 min before hypoxia.
Measurement of ROS generation
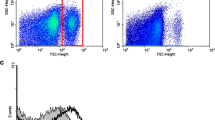
Cells were incubated with 2 mM DCFHDA–AM (Sigma, #287810) for 20 min at 37 °C in a 5 % CO2 incubator, washed and resuspended in PBS at 1 × 106 cells/ml. Cells were analyzed by flow cytometry (Becton–Dickinson) at an excitation wavelength of 514 nm, and the fluorescence intensity of DCF was measured at an emission wavelength of 525 nm. Untreated cells served as controls. The amount of intracellular ROS was expressed as the fold-increase of DCF fluorescence compared with the control [16]. The experiments were repeated three times.
Xanthine Oxidase Activity Assay
XO activity assay was performed according to the manufacturer’s instructions (Xanthine Oxidase Assay Kit; BioVision, #K710-100). With reagents provided in the kit, we generated a H2O2 standard curve and used it to calculate the H2O2 produced by various concentrations of XO. 1 × 106 cells were homogenized in cold assay buffer and lysates were centrifuged at 12,000×g for 15 min, the supernatants collected and protein concentration determined by BCA protein assay (PIERCE, #23225). Cellular extracts (30 μg) were then incubated in a 96-well plate with reagents provided in the kit. In the assay, XO oxidizes xanthine to H2O2 which reacts stoichiometrically with OxiRed™ probe to generate color (λ = 570 nm). Enzyme activity was determined in 100 μl reaction volume spectrophotometrically (λ = 570 nm). Relative XO activity was calculated as a ratio of emission of treated cells to untreated cells [17]. The experiments were repeated three times.
Measurement of intracellular Ca2+
After experimentation, cells were loaded with fluo-3 AM (Invitrogen, #F23915) in 1 % working solution at 37 °C for 30 min, washed three times with Ca2+-free PBS to remove extracellular fluo-3 AM, and resuspended in PBS at a concentration of 1 × 106 cells/ml. Cells were analyzed by flow cytometry (Becton–Dickinson) at an excitation wavelength of 488 nm, and the fluorescence intensity was measured at an emission wavelength of 530 nm [18]. Untreated cells served as controls. The amount of intracellular Ca2+ [(Ca2+)i] was expressed as the fold-increase of fluorescence compared with the control. The experiments were repeated three times.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted from CMECs using TRIzol reagent (Life Technologies), the purity of the samples was assessed determining the 260/280 nm ratio. Reverse transcription was performed using the PrimeScript reverse transcriptase (Takara Bio, #2680A), according to the manufacturer’s instructions. The cDNAs were prepared from 1 μg of total RNA. Reverse transcription conditions comprised an initial incubation step at 25 °C for 10 min to allow random hexamers annealing, followed by cDNA synthesis at 37 °C for 120 min, and final inactivation step for 5 min at 95 °C. Real-time-PCR was performed in a total reaction volume of 20 μl with Platinum SYBRGreen PCR SuperMix-UDG (Life Technologies) using the Eco™ Real-Time PCR Detection System (illumine, San Diego, CA, USA) following the manufacturer’s protocol. The thermal cycling protocol was as follows: denaturation at 94 °C for 0.5 min, followed by 40 cycles at 95 °C for 30 s, annealing at 60 °C for 0.5 min, and extension at 72 °C for 1 min. GAPDH mRNA was amplified as an internal control. The primers used were as follows: The primers used were 5′-caagattgtcagcaatgcatcc-3′(sense) and 5′-atcacgccacagctttccagag-3′(antisense) for rat XDH; 5′-gtgtgacctggatccaggcttc-3′(sense) and 5′-ttcagttcagggatccaggctg-3′(antisense) for rat GAPDH. For each sample, the expression of the XDH gene was normalized with the GAPDH mRNA content. The experiments were repeated three times.
Western blotting analysis
CMECs were homogenized in RIPA lysis buffer (Beyotime, #P0013C) containing 1 × Phosphatase Inhibitor Cocktail (Cell Signaling Technology, #5870S) and 1 μg/ml each of aprotinin (Sigma, #A6106) and leupeptin (Sigma, #L2884). Aliquots of 40 μg of protein from each sample were boiled in sodium dodecylsulfate buffer (Beyotime, #P0015) for 5 min. The protein was separated by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes, and probed with antibodies for XO (Epitomics, #ab109235). The primary antibodies at the concentration of 1:1,000 were exposed for overnight at 4 °C. Next, horseradish peroxidase-conjugated secondary antibodies (Beyotime, #A0208) at the concentration of 1:5,000 were added, and incubated for 1 h at 37 °C. The membranes were then developed by enhanced chemiluminescence (Beyotime, #P0018). The same membranes were reprobed with antibody for Tubulin (Beyotime, #AT819). The blotting film was quantified using a scanner and a densitometry program (Image J).
Cell apoptosis
Cell apoptosis was assessed using Annexin V-FITC/PI Kit (Roche, #1185877001 Basel Switzerland) and Caspase 3 Activity Assay Kit (Beyotime, #C1116, Jiangsu, China). Briefly, 1 × 106 cells were collected and resuspended in 100 μl medium buffer. 2 μl of Annexin V and 2 μl of PI solution were added to the cell suspending solution and incubated for 15 min preventing from the light at room temperature. Then, the suspensions were analyzed by a flow cytometric machine immediately. Ten thousand events were acquired on a FACSCalibur (Becton–Dickinson, Mountainview, CA) and analyzed with Cell Quest (Becton–Dickinson, Mountainview, CA) software [19]. The experiments were repeated three times.
In caspase 3 activity assay, cell lysates were prepared by incubating 2 × 106 cells/ml in extraction buffer (25 mM Tris–HCl, pH 7.5, 20 mM MgCl2, and 150 mM NaCl, 1 % Triton X-100, 25 μg/ml leupeptin, and 25 μg/ml aprotinin) for 30 min on ice. Lysates were centrifuged at 12,000×g for 15 min, the supernatants were collected and protein concentration was determined by BCA protein assay (Pierce, #23227, Rockford, IL). Cellular extracts (30 μg) were then incubated in a 96-well microtiter plate with 20 ng Ac-DEVD-pNA for 2 h at 37 °C. Caspase 3 activity was measured by cleavage of the Ac-DEVD-pNA to pNA, the absorbance of which was measured spectrophotometrically (λ = 405 nm). Relative caspase 3 activity was calculated as a ratio of emission of treated cells to untreated cells [20]. The experiments were repeated three times.
Statistical analysis
The statistic software package SPSS 13.0 was used for analysis of data. Statistical comparisons were performed using the paired, two-tailed Student’s t test for experiments consisting of two groups only and with one-way ANOVA and followed by SNK analysis for experiments consisting of more than two groups. Results were considered statistically significant when p < 0.05. Data are presented as mean ± SE.
Results
Effect of HGF on ROS production induced by H/R in CMECs
In the present study, we demonstrated that H/R promoted cellular ROS production in CMECs. The changes in ROS production were detected by DCFHDA–AM staining. Pretreatment of CMECs with HGF (10 and 20 ng/ml) before H/R treatment was capable of preventing cellular ROS increase, as assessed by flow cytometry analysis (Fig. 1a). In addition, the inhibition of cellular ROS increase by HGF was in a dose-dependent manner, as shown in (Fig. 1b).

HGF attenuates H/R-induced ROS production. a Representative flow cytometry analysis of DCFHDA staining CMECs (λ = 588 nm) of HGF 10 ng/ml pretreatment group and HGF 20 ng/ml pretreatment group, and compared with the H/R group. b Summary data (n = 3 biological replicates in all experimental groups) of flow cytometry analysis of DCFHDA staining CMECs (λ = 588 nm) of HGF 10 ng/ml pretreatment group and HGF 20 ng/ml pretreatment group, and compared with the H/R group *p < 0.05 versus H/R group
Role of XO on ROS production induced by H/R in CMECs
Endogenous ROS signaling was investigated in CMECs using DCFHDA–AM dye, which becomes fluorescent on oxidation to DCF in the cell. H/R treatment increased intracellular DCF fluorescence compared with normoxia (control group). But the increased production of ROS after H/R was significantly attenuated when the cells were pretreated for 30 min before hypoxia with allopurinol (20 μmol/L, 40 μmol/L), an inhibitor of XO (Fig. 2a, b).

Inhibition of XO attenuates H/R-induced ROS production. a Representative flow cytometry analysis of DCFHDA staining CMECs (λ = 588 nm) after H/R. Allopurinol pretreatment group (20, 40 μM) compared with the H/R group. b Summary data (n = 3 biological replicates in all experimental groups) of flow cytometry analysis of DCFHDA staining CMECs (λ = 588 nm) after H/R. Allopurinol pretreatment group (20, 40 μM) compared with the H/R group *p < 0.05 versus H/R group
Effect of HGF on XO under H/R conditions in CMECs
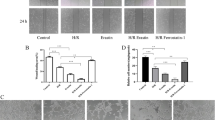
XDH mRNA was qualified by qRT-PCR. We found that after H/R, XDH mRNA significantly increased, indicating a de novo synthesis of XOR after H/R. However, pretreatment of HGF showed no significant effect on the increase of XDH mRNA (Fig. 3a). XO protein levels were detected by Western blot, and it showed that XO protein level increased after H/R. Besides, XO activity detected by Xanthine Oxidase Assay Kit increased after H/R, which agreed with the changes of XO protein levels, XDH mRNA, cellular ROS production after H/R. Pretreatment of HGF blocked the increase of both XO protein (Fig. 3b, c) and XO activity (Fig. 3d).

Effect of HGF on XDH transcription, XO protein level and XO activity changes after H/R. a RT-PCR analysis of XDH mRNA after H/R. HGF pretreatment group (10 ng/ml, 20 ng/ml) compared with the H/R group. The level of GAPDH mRNA is used as a control marker. XDH mRNA increased after H/R and HGF showed no significant effect on the increase of XDH mRNA. n = 3 biological replicates in all experimental groups. b Representative immunoblot for XO and GAPDH in CMECs after H/R. HGF pretreatment group (10 ng/ml, 20 ng/ml) compared with the H/R group. In CMECs, XO protein levels increased after H/R. Pretreatment of HGF blocked the increase of XO protein induced by H/R. c Summary data (n = 3 biological replicates in all experimental groups.) of immunoblot for XO and GAPDH in CMECs after H/R. HGF pretreatment group (10, 20 ng/ml) compared with the H/R group. *p < 0.05 versus H/R group. d XO activity assayed with Xanthine Oxidase Assay Kit in CMECs after H/R. HGF pretreatment group (10, 20 ng/ml) compared with the H/R group. n = 3 biological replicates in all experimental groups. *p < 0.05 versus H/R group
Effect of HGF on cytosolic Ca2+ concentration under H/R conditions in CMECs
Cytosolic Ca2+ concentration increased after H/R and HGF attenuated this increase in a dose-dependent manner (Fig. 4a, b). To testify the role of cytosolic Ca2+ concentration elevation on XO protein level and XO activity after H/R, we pretreated CMECs with the cell-permeable Ca2+ chelator BAPTA-AM (1 μM). We found that BAPTA-AM (1 μM) partially prevented the increase in XO protein expression after H/R (Fig. 4c, d). The increase in XO activity after H/R was also attenuated by BAPTA-AM (1 μM) (Fig. 4e).

HGF suppresses XO activation and protein level elevation by attenuating increase of cytosolic Ca2+ concentration induced by H/R. a Representative flow cytometry analysis of fluo-3 staining CMECs (λ = 588 nm) after H/R. HGF pretreatment group (10, 20 ng/ml) compared with the H/R group. Cytosolic Ca2+ concentration increased after H/R, and HGF attenuated the increase of cytosolic Ca2+ concentration induced by H/R. b Summary data (n = 3 biological replicates in all experimental groups) of flow cytometry analysis of fluo-3 staining CMECs (λ = 588 nm) HGF pretreatment group (10, 20 ng/ml) compared with the H/R group. *p < 0.05 versus H/R group. *p < 0.05 versus H/R group. c Representative immunoblot for XO and GAPDH in CMECs after H/R. A Ca2+ chelator, BAPTA-AM (1 μM)-treated group was compared with H/R group. In CMECs, BAPTA-AM (1 μM) significantly reduced XO protein levels at H4R1. *p < 0.05 versus H/R group. d Summary data (n = 3 biological replicates in all experimental groups.) of immunoblot for XO and GAPDH in CMECs after H/R. BAPTA-AM (1 μM)-treated group was compared with H/R group. *p < 0.05 versus H/R group. e XO activity assayed with Xanthine Oxidase Assay Kit in CMECs after H/R. BAPTA-AM (1 μM)-treated group was compared with H/R group. n = 3 biological replicates in all experimental groups. In CMECs, BAPTA-AM (1 μM) significantly reduced XO activity after H/R. *p < 0.05 versus H/R group
Role of XO on H/R-induced cell apoptosis in CMECs
To investigate H/R-induced CMECs apoptosis, we used flow cytometric analysis with Annexin V-FITC/PI double staining and caspase 3 activity assays. When CMECs were pretreated with allopurinol (20, 40 μmol/L) for 30 min before hypoxia, allopurinol significantly protected CMECs from apoptosis (Fig. 5a, b). Given that caspase 3 plays a crucial role in cell apoptosis, we also found that allopurinol inhibited caspase 3 activation after H/R (Fig. 5c).

Inhibition of XO attenuated H/R-induced CMECs apoptosis. a Representative Annexin V-FITC/PI double staining CMECs were analyzed by flow cytometry. Allopurinol treatment group (20, 40 μM) compared with the H/R group. Allopurinol inhibited CMECs apoptosis induced by H/R. b Summary data (n = 3 biological replicates in all experimental groups.) of Annexin V-FITC/PI double staining CMECs were analyzed by flow cytometry. Allopurinol treatment group (20, 40 μM) compared with the H/R group. *p<0.05 versus H/R group. c Caspase 3 activity within CMECs was assayed. Allopurinol treatment group (20, 40 μM) compared with the H/R group. *p<0.05 versus H/R group. Allopurinol inhibited increased caspase 3 activity in CMECs induced by H/R. n = 3 biological replicates in all experimental groups
Effect of HGF on H/R-induced cell apoptosis in CMECs
Cell apoptosis was investigated by flow cytometric analysis with Annexin V-FITC/PI double staining and caspase 3 activity assays. We found that after H/R, cell apoptosis rate significantly increased, pretreatment of HGF inhibited H/R-induced apoptosis in CMECs (Fig. 6a, b). Caspase 3 activity was measured by cleavage of the Ac-DEVD-pNA to pNA, the absorbance of which was measured spectrophotometrically (λ = 405 nm). Pretreatment of HGF blocked the increase of caspase 3 activity after H/R (Fig. 6c).

HGF attenuated H/R-induced CMECs apoptosis. a Representative Annexin V-FITC/PI double staining CMECs were analyzed by flow cytometry. HGF treatment group (10, 20 ng/ml) compared with the H/R group. b Summary data (n = 3 biological replicates in all experimental groups.) of Annexin V-FITC/PI double staining CMECs were analyzed by flow cytometry. HGF treatment group (10, 20 ng/ml) compared with the H/R group. **p<0.01 versus H/R group. c Caspase 3 activity within CMECs was assayed. HGF treatment group (10, 20 ng/ml) compared with the H/R group. **p<0.01 versus H/R group. HGF inhibited increased caspase 3 activity in CMECs induced by H/R. n = 3 biological replicates in all experimental groups
Discussion
H/R, which generates cytotoxic ROS and promotes the recruitment of inflammatory leukocytes, induces cell dysfunction and cell death. Endothelial cells appear to be the first cell type injured by ROS generated during H/R [7]. Reduction of oxidative stress improves coronary microvasculature [22]. In the previous study, HGF function in a unique fashion in that it does not employ ROS as its own downstream signaling molecules, but rather inhibits one of the cellular machineries responsible for ROS generation, such as Rac-1 [23]. XO, one of the ROS-generating enzymes, has been found to be activated in endothelial cells by hypoxia [24]. In the present study, we extended to clarify the mechanisms by which HGF inhibits H/R-induced XO activation and oxidative stress in endothelial cells.
Consistent with previous study [23], we found that H/R induced a significant increase of ROS generation in CMECs. However, potential sources of ROS include XO [4], NADPH oxidase [25], the metabolic cascade of arachidonic acid [26], and the mitochondrial respiratory chain [27]. Under H/R conditions, XO is found to be the major source of ROS in rats pulmonary circulation [28] and rats jugular venous [2]. However, mitochondrial respiratory chain is found to be the major source of ROS in HUVECs [29] and in embryonic chick cardiomyocytes [27]. To identify the major source of ROS in H/R-treated CMECs, we pretreated the cell before hypoxia with allopurinol, a potent XO inhibitor, and found that cellular ROS increase is blocked by about 80 %. Allopurinol also significantly ameliorates H/R-induced CMECs apoptosis. Our results show that XO accounts for largely the increase of cellular ROS induced by H/R in CMECs.
In our present study, we found that pretreatment with HGF before hypoxia significantly blocked the cellular ROS increase induced by H/R. Since XO accounts for largely the increase of cellular ROS induced by H/R in CMECs. We hypothesized that HGF reduced ROS production by affecting the activation of XO. Of note, XO is first synthesized as XDH, an XO precursor, and needs to be converted to be XO to produce ROS. So we tested the effect of HGF on the changes of XDH mRNA, XO protein levels and XO activities under H/R conditions. XDH mRNA, XO protein levels and XO activities all increased under H/R conditions. These data illustrate a de novo synthesis and activation after H/R, and further confirm that XO is the major source of ROS after H/R in CMECs. Pretreatment of HGF before hypoxia attenuated the increase of XO protein levels and XO activities. However, HGF has no effect on the increase of XDH mRNA. Thus, HGF may not exert the regulation on XO at the transcriptional level, and may regulate XO at the translational or posttranslational levels. In our study, we found that HGF protects CMECs from H/R-induced cell apoptosis. Since HGF suppresses XO activation and allopurinol also ameliorates H/R-induced CMECs apoptosis, HGF may protect CMECs from H/R-induced injury, at least in part by reducing XO production and activation.
Xanthine oxidase is identified to be regulated by Ca2+ [21], and an unidentified Ca2+-dependent protease is believed to be involved in the cleavage of XDH to XO. Our present study detects that cytosolic Ca2+ concentration increases after H/R. This agrees with the former study that reperfusion enhanced cytosolic Ca2+ overload via Ca2+ influx and Ca2+ release from the endoplasmic reticulum [30]. Pretreatment CMECS with BAPTA-AM, a cell-permeable Ca2+ chelator, significantly blocks the increase of XO protein level and XO activity. These data suggest that increased cytosolic Ca2+ concentration after H/R plays a crucial role in the increases of both XO protein level and XO activity. Furthermore, we tested the effect of HGF on cytosolic Ca2+ concentration under H/R conditions. Pretreatment of HGF before hypoxia attenuated the increase of cytosolic Ca2+ concentration induced by H/R. These data suggest that HGF regulates XO protein level and XO activity at the posttranslational by inhibiting the increase of cytosolic Ca2+ concentration under H/R conditions. Recent studies also reported that intermittent hypoxia increased trypsin-like endoprotease activity and activated XO by increased proteolytic conversion of XDH to XO [31]. A novel protease that cleaves XDH to XO has been found in the mitochondrial intermembrane space [32], but whether this protease is involved in the HGF regulation of XO is beyond the scope of our present study.
In conclusion, these findings suggest a novel mechanism whereby HGF protects CMECs from H/R-induced oxidative stresses. H/R induces a de novo synthesis of XDH, the XO precursor. In addition, H/R increases cytosolic Ca2+ concentration and promotes a Ca2+-involved XO conversion and XO activation. XO accounts for the increase of ROS induced by H/R. HGF has no effect on XDH mRNA, but inhibits the increase of cytosolic Ca2+ concentration induced by H/R. Thereby HGF regulates the XO protein levels and XO activity at the posttranslational level under H/R conditions. Thus, HGF protects CMECs from H/R-induced oxidative stresses and H/R-induced injury.
References
Beetsch JW, Park TS, Dugan LL, Shah AR, Gidday JM (1998) Xanthine oxidase-derived superoxide causes reoxygenation injury of ischemic cerebral endothelial cells. Brain Res 786:89–95
Ono T, Tsuruta R, Fujita M, Aki HS, Kutsuna S, Kawamura Y, Wakatsuki J, Aoki T, Kobayashi C, Kasaoka S, Maruyama I, Yuasa M, Maekawa T (2009) Xanthine oxidase is one of the major sources of superoxide anion radicals in blood after reperfusion in rats with forebrain ischemia/reperfusion. Brain Res 1305:158–167
Ali OS, Abdelgawad HM, Mohammed MS, El-Awady RR (2013) Ischemic heart diseases in Egypt: role of xanthine oxidase system and ischemia-modified albumin. Heart Vessels. doi:10.1007/s00380-00013-00413-00383
Meneshian A, Bulkley GB (2002) The physiology of endothelial xanthine oxidase: from urate catabolism to reperfusion injury to inflammatory signal transduction. Microcirculation 9:161–175
Agarwal A, Banerjee A, Banerjee UC (2011) Xanthine oxidoreductase: a journey from purine metabolism to cardiovascular excitation-contraction coupling. Crit Rev Biotechnol 31:264–280
Hare JM, Berry CE (2004) Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol 555:589–606
Wang X, Zhou Y, Kim HP, Song R, Zarnegar R, Ryter SW, Choi AMK (2004) Hepatocyte Growth Factor Protects against Hypoxia/Reoxygenation induced Apoptosis in Endothelial Cells. J Biol Chem 279:5237–5243
Date I, Takaga N, Takagi K, Kago T, Matsumoto K, Nakamura T, Takeo S (2004) Hepatocyte growth factor attenuates cerebral ischemia-induced learning dysfunction. Biochem Biophys Res Commun 319:1152–1158
Makiuchi A, Yamaura K, Mizuno S, Matsumoto K, Nakamura T, Amano J, Ito Ken-ichi (2007) Hepatocyte growth factor prevents pulmonary ischemia-reperfusion injury in mice. J Heart Lung Transplant 26:935–943
Guo Y, Su L, Li Y, Guo N, Xie L, Zhang D, Zhang X, Li H, Zhang G, Wang Y, Liu C (2013) The synergistic therapeutic effect of hepatocyte growth factor and granulocyte colony-stimulating factor on pulmonary hypertension in rats. Heart Vessels. doi:10.1007/s00380-013-0395-1
Li H, Yu H, Zhao J, Huang Y, Chen Q, Zhang N (2010) HGF suppresses high glucose-mediated oxidative stress in mesangial cells by activation of PKG and inhibition of PKA. Free Radic Biol Med 49:467–473
Zhou YJ, Yang HW, Wang XG, Zhang H (2009) Hepatocyte growth factor prevents advanced glycation end products-induced injury and oxidative stress through a PI3 K/Akt-dependent pathway in human endothelial cells. Life Sci 85:670–677
Nishida M, Carley WW, Gerritsen ME, Ellingsen O, Kelly RA, Smith TW (1993) Isolation and characterization of human and rat cardiac microvascular endothelial cells. Am J Physiol Heart Circ Physiol 264:H639–H652
Zhang Z, Li W, Sun D, Zhao L, Zhang R, Wang Y, Zhou X, Wang H, Cao F (2011) Toll-like receptor 4 signaling in dysfunction of cardiac microvascular endothelial cells under hypoxia/reoxygenation. Inflamm Res 60:37–45
Yu G, Bolon M, Laird DW, Tyml K (2010) Hypoxia and reoxygenation-induced oxidant production increase in microvascular endothelial cells depends on connexin40. Free Radic Biol Med 49:1008–1013
Kong R, Jia G, Cheng ZX, Wang YW, Mu M, Wang XSJ, Pan SH, Gao Y, Jiang HC, Dl Dong, Sun B (2012) Dihydroartemisinin enhances Apo2L/TRAIL-mediated apoptosis in pancreatic cancer cells via ROS-mediated Up-regulation of death receptor 5. PLoS ONE 7:e37222
Ha Y, Dun Y, Thangaraju M, Duplantier J, Dong Z, Liu K, Ganapathy V, Smith SB (2011) Sigma receptor 1 modulates endoplasmic reticulum stress in retinal neurons. Invest Ophthalmol Vis Sci 52:527–540
Przygodzkia T, Sokal A, Bryszewska M (2005) Calcium ionophore A23187 action on cardiac myocytes is accompanied by enhanced production of reactive oxygen species. Biochim Biophys Acta 1740:481–488
Piekarska J, Szczypka M, Mrukowicz BO, Gorczykowski M (2009) Effect of phytohaemagglutinin-P on apoptosis and necrosis in Trichinella spiralis infected mice. Vet Parasitol 159:240–244
Hu R, Zhou P, Peng YB, Xu X, Ma J, Liu Q, Zhang L, Wen XD, Qi LW, Gao N, Li P (2012) 6-Shogaol induces apoptosis in human hepatocellular carcinoma cells and exhibits anti-tumor activity in vivo through endoplasmic reticulum stress. PLoS One 7:e39664
McNally JS, Saxena A, Cai H, Dikalov S, Harrison DG (2005) Regulation of xanthine oxidoreductase protein expression by hydrogen peroxide and calcium. Arterioscler Thromb Vasc Biol 25:1623–1628
Shida T, Nozawa T, Sobajima M, Ihori H, Matsuki A, Inoue H (2013) Fluvastatin-induced reduction of oxidative stress ameliorates diabetic cardiomyopathy in association with improving coronary microvasculature. Heart Vessels. doi:10.1007/s00380-013-0402-6
Ozaki M, Haga S, Zhang HQ, Irani K, Suzuki S (2003) Inhibition of hypoxia/reoxygenation-induced oxidative stress in HGF-stimulated antiapoptotic signaling: role of PI3-K and Akt kinase upon rac1. Cell Death Differ 10:508–515
Wang G, Qian P, Jackson FR, Qiana G, Wu G (2008) Sequential activation of JAKs, STATs and xanthine dehydrogenase/oxidase by hypoxia in lung microvascular endothelial cells. Int J Biochem Cell Biol 40:461–470
Peng T, Yu G, Feng Q, Tyml K (2007) Abrupt reoxygenation of microvascular endothelial cells after hypoxia activates ERK1/2 and JNK1, leading to NADPH oxidase-dependent oxidant production. Microcirculation 14:125–136
Krause GS, White BC, Aust S, Nayini NR, Kumar K (1988) Brain cell death following ischemia and reperfusion: a proposed biochemical sequence. Crit Care Med 16:714–726
Kondapalli J, Loor G, Iwase H, Chandel NS, Waypa GB, Guzy RD, Hoek TLV, Schumacker PT (2011) Mitochondrial oxidant stress triggers cell death in simulated ischemia–reperfusion. Biochim Biophys Acta 1813:1382–1394
Ogawa J, Saito S, Minamiya Y (2005) Pulmonary reexpansion causes xanthine oxidase-induced apoptosis in rat lung. Am J Physiol 289:L400–l406
Laemmel E, Matharan ST, Duranteau J, Vicaut E (2004) Reoxygenation after hypoxia and glucose depletion causes reactive oxygen species production by mitochondria in HUVEC. Am J Physiol Heart Circ Physiol 287:R1037–R1043
Peters SC, Piper HM (2007) Reoxygenation-induced Ca2+ rise is mediated via Ca2+ influx and Ca2+ release from the endoplasmic reticulum in cardiac endothelial cells. Cardiovasc Res 73:164–171
Nanduri J, Vaddi DR, Khan SA, Wang N, Makerenko V, Prabhakar NR (2013) Xanthine oxidase mediates hypoxia-inducible factor-2a degradation by intermittent hypoxia. PLoS One 8:e75838
Lapatto R, Saksela M, Raivio KO (1999) Irreversible conversion of xanthine dehydrogenase to xanthine oxidase by a mitochondrial protease. FEBS Lett 433:117–120
Acknowledgments
This work was supported by grants (81070185, 81102079) from the National Natural Science Foundation of China.
Conflict of interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhang, Y., Hu, S. & Chen, Y. Hepatocyte growth factor suppresses hypoxia/reoxygenation-induced XO activation in cardiac microvascular endothelial cells. Heart Vessels 30, 534–544 (2015). https://doi.org/10.1007/s00380-014-0547-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00380-014-0547-y