
Abstract
A 5-year-old girl developed cardiopulmonary arrest after crying. From the electrocardiogram and echocardiography, a left ventricular noncompaction (LVNC) with long QT syndrome (LQT) was suspected as the cause of the cardiopulmonary arrest, and treatment with a β-blocker and a calcium antagonist was then begun. A genetic screening of LQT-related genes revealed a previously reported heterozygous KCNQ1 mutation. The association of LVNC and LQT is an extremely rare condition, and long-term treatment based on the characteristics of both disorders is required. Also, the association of cardiomyopathy and LQT could become a new clinical entity in the future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Long QT syndrome (LQT) is a group of ion-channel disorders of the myocardium that may prolong the repolarization of the cardiac cycle [1]. According to the genotype investigation, 12 subtypes (LQT1–12) have been reported [2]; each subtype has its own clinical characteristics, and the treatment strategy differs for each subtype. Long QT syndrome is known as the most important cause of sudden cardiac death in the young [3], and may mostly result from the occurrence of ventricular fibrillation (VF) or torsade de pointes (TdP).
Here we report the case of a girl with left ventricular noncompaction (LVNC) and LQT, which were confirmed after resuscitation from cardiopulmonary arrest.
Case report
A 5-year-old girl had syncope after intense crying at her kindergarten. Her mother noticed cyanosis around her lips and then she developed cardiopulmonary arrest. Bystander cardiopulmonary resuscitation (CPR) was started immediately by a kindergarten teacher, who called for an ambulance. An automated external defibrillator (AED) revealed pulseless electrical activity, and there was no indication for defibrillation. She was transported to our hospital under continuous CPR.
She had been followed up for a diagnosis of epilepsy after two episodes of afebrile convulsions when she was 3 years old. She had a syncopal attack during the follow-up period, and multifocal spike waves were noted on the electroencephalogram. She had been administered carbamazepine since then, after which the spike waves disappeared during the follow-up period. Except for an episode of afebrile convulsions at age 4 years, she did not experience any further episodes of convulsions or syncope. Magnetic resonance imaging revealed no brain abnormalities.
On arrival at our hospital, sinus rhythm had resumed; however, she required intubation and respiratory support for her respiratory failure and cardiac dysfunction. Her cardiac function then improved gradually, but about 6 h after arrival, TdP and VT emerged in the intensive care unit. While we were preparing to defibrillate her, performing cardiac compressions for about 1 min, sinus rhythm resumed spontaneously (Fig. 1) and her cardiac function improved.

Monitor recording obtained while the patient was in the intensive care unit. Upper panel occurrence of torsade de pointes (TdP), which terminated within several beats. Lower panel occurrence of long-lasting TdP or ventricular fibrillation
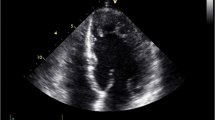
The electrocardiogram obtained after the CPR exhibited a prolonged QTc interval (Fig. 2, QTc = 0.6 s), and the patient was suspected as having LQT. There were no electrolyte imbalances at the time of hospitalization. Her cardiac function improved gradually after CPR. An echocardiogram revealed a spongy dysplastic left ventricular myocardium with prominent trabeculations and deep recesses, indicating LVNC (Fig. 3). We therefore started the patient on propranolol and verapamil to control her VT. Her respiratory support was discontinued 3 days after hospitalization. After administration of propranolol and verapamil, TdT and VT no longer emerged.

Electrocardiogram recorded after the resuscitation. The electrocardiogram after the resuscitation showed normal sinus rhythm with left QRS axis deviation (−15°). The QTc interval was prolonged to 0.6 s. There were also flattened T waves in the left precordial leads (V5 and V6)

Echocardiogram. Prominent trabeculations and a deep recess were detected
Electrocardiographic examinations and a genetic screening of LQT-related genes were performed on the patient, her sister and brother, her parents, her paternal and maternal grandfathers and grandmothers, and a maternal uncle (Fig. 4). She, her brother, her father, and her paternal grandmother were found to have a previously reported heterozygous KCNQ1 mutation c.1831 G > T in exon 15 (p. D611T). No prolongation of QT intervals or echocardiographic abnormalities were found in family members (her brother, her father, and her paternal grandmother) who had a KCNQ1 mutation. Although her development had been normal until this event, the patient manifested mild mental retardation because of ischemic brain damage. She underwent rehabilitation and attended a school for handicapped children with a restriction on swimming.

Family tree of our patient. The arrow shows the proband. KCNQ1 mutations were detected in the proband, and her brother, father, and grandmother
Discussion
Left ventricular noncompaction is a congenital cardiomyopathy with a spongy morphological appearance and deep intertrabecular sinusoids in communication with the ventricular cavity [4]. The diagnosis is mainly made by two-dimensional echocardiography, cardiac magnetic resonance imaging, or left ventricular angiography.
The echocardiogram reveals that prominent trabeculations and deep recesses are noted in the ventricular myocardium [5]. However, so far there has been no distinct definition of LVNC [6]. Koh et al. [7] reported that a left ventricular myocardial deformation is reduced in the longitudinal and circumferential dimensions and manifests with tight systolic–diastolic coupling in children with LVNC.
Genetic mutations were first reported in the G4.5 gene in patients with an isolated LVNC [8]. Z-line and mitochondrial mutations and X-linked inheritance resulting from mutations in the G4.5 gene encoding tafazzin could be a pathogenesis for the disease. In this report, the gene defect differed among the families, and thus there did not appear to be any obvious genotype–phenotype correlation that would allow for the differentiation of the clinical course to be predicted. In addition, the cardiac phenotypes that occur as a result of G4.5 mutations may vary significantly. On the other hand, in patients with LVNC associated with congenital heart disease, a so-called nonisolated LVNC, mutations in the α-dystrobrevin gene have been reported [9]. In this report, the α-dystrobrevin mutation resulted in a phenotype of a dilated hypertrophic cardiomyopathy with deep trabeculations associated with congenital heart disease, consistent with the criteria for LVNC. However, the phenotype in this family had considerable variability. Consequently, the details of the relation between an ion-channel dysfunction and the maldevelopment of the ventricular myocardium are not well described. Further genetic studies are needed to discover whether a combined mutation with G4.5 or α-dystrobrevin and KCNQ1 could have contributed to this clinical manifestation in this patient.
The association of LVNC with LQT is extremely rare. SCN5A mutations are frequently associated with LVNC. However, only 2 of 62 patients were found to have LQT in SCN5A-positive LVNC [10]. SCN5A mutations are well known as a cause of LQT3 syndrome and Brugada syndrome. Ogawa et al. [11] reported a KCNH2 mutation in two patients with LQT and LVNC. KCNH2 mutations have been known to be the cause of LQT2 syndrome.
KCNQ1 mutations are known as a cause of LQT [12]. However, to the best of our knowledge there have been no previous reports on the association of a KCNQ1 mutation and LVNC, so this is the first report suggesting an association between LQT1 and LVNC.
The association of cardiomyopathy and LQT could become a new clinical entity in the future. In 2006, the American Heart Association scientific statement on the classification of cardiomyopathies formally classified LVNC as its own disease entity, as a primary cardiomyopathy with a genetic origin, in the same category as ion-channel disorders [13]. Long-term follow-up will be required to reveal further associations between both disorders.
Conclusion
The association of LQT with LVNC is extremely rare. There have been only two patients with SCN5A mutations and two patients with KCNH2 mutations reported to date. This is the first report of a KCNQ1 mutation with LQT and LVNC. A genetic screening of LQT-related genes is recommended for patients with a long QT interval and LVNC.
References
Moss AJ (2003) Long QT syndrome. JAMA 289:2041–2044
Hedley PL, Jørgensen P, Schlamowitz S, Wangari R, Moolman-Smook J, Brink PA, Kanters JK, Corfield VA, Christiansen M (2009) The genetic basis of long QT and short QT syndromes: a mutation update. Hum Mutat 30:1486–1511
Li H, Fuentes-Garcia J, Towbin JA (2000) Current concepts in long QT syndrome. Pediatr Cardiol 21:542–550
Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB (2006) Contemporary definitions and classification of the cardiomyopathies. Circulation 113:1807–1816
Jenni R, Oechslin E, Schneider J, Attenhofer Jost C, Kaufmann PA (2001) Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. Heart 86:666–671
Chin TK, Perloff JK, Williams RG, Jue K, Mohrmann R (1990) Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circulation 82:507–513
Koh C, Hong WJ, Wong SJ, Cheung YF (2010) Systolic–diastolic coupling of myocardial deformation of the left ventricle in children with left ventricular noncompaction. Heart Vessels 25:493–499
Bleyl SB, Mumford BR, Thompson V, Carey JC, Pysher TJ, Chin TK, Ward K (1997) Neonatal, lethal noncompaction of the left ventricular myocardium is allelic with Barth syndrome. Am J Hum Genet 61:868–872
Ichida F, Tsubata S, Bowles KR, Haneda N, Uese K, Miyawaki T, Dreyer WJ, Messina J, Li H, Bowles NE, Towbin JA (2001) Novel gene mutations in patients with left ventricular noncompaction or Barth syndrome. Circulation 103:1256–1263
Shan L, Makita N, Xing Y, Watanabe S, Futatani T, Ye F, Saito K, Ibuki K, Watanabe K, Hirono K, Uese K, Ichida F, Miyawaki T, Origasa H, Bowles NE, Towbin JA (2008) SCN5A variants in Japanese patients with left ventricular noncompaction and arrhythmia. Mol Genet Metab 93:468–474
Ogawa K, Nakamura Y, Terano K, Ando T, Hishitani T, Hoshino K (2009) Isolated non-compaction of the ventricular myocardium associated with long QT syndrome: a report of 2 cases. Circ J 73:2169–2172
Bokil NJ, Baisden JM, Radford DJ, Summers KM (2010) Molecular genetics of long QT syndrome. Mol Genet Metab 101:1–8
Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB; American Heart Association; Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; Council on Epidemiology and Prevention (2006) Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee. Circulation 113:1807–1816
Acknowledgments
The authors thank Mr. John Martin for his linguistic assistance with this article.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nakashima, K., Kusakawa, I., Yamamoto, T. et al. A left ventricular noncompaction in a patient with long QT syndrome caused by a KCNQ1 mutation: a case report. Heart Vessels 28, 126–129 (2013). https://doi.org/10.1007/s00380-012-0235-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00380-012-0235-8