
Abstract
We assessed the diversity of arbuscular mycorrhizal (AM) fungi colonizing maize roots grown in a long-term monoculture experiment established at Martonvásár, Hungary, 50 years ago to understand the effect of this extremely long monoculture on the community structure of these organisms. Mycorrhizal colonization of root samples was analyzed by PCR amplification and sequencing of partial ribosomal small subunit DNA fragments of fungal origin. Of the 257 sequences recovered, 203 belonged to Glomeromycota AM fungi. Phylogenetic analysis assigned the Glomeromycota sequences into 22 operational taxonomic units belonging to three families including Archaeosporaceae, Glomeraceae, and Paraglomeraceae. In agreement with previous reports, Glomus group A fungi dominated the arbuscular mycorrhizal fungi community of maize, but we found a relatively high richness of phylotypes within this group even after such an extreme and durable reduction of host plant diversity. Agricultural practices, including mineral fertilization and incorporating stalk residues, significantly affected the diversity within Glomus group A.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Arbuscular mycorrhizal fungi (AMF) influence ecosystem processes (Maherali and Klironomos 2007), have a role in maintaining plant diversity in natural communities, induce a variety of growth responses in co-existing plant species (van der Heijden et al. 1998), and, due to their enzymatic activity, they contribute to organic matter cycling (Theuri and Buscot 2010). They have been shown to increase the productivity of most crop species including maize (Sylvia et al. 1993). AM fungi enhance the uptake of phosphorus, but they also contribute to the absorption of other immobile ions, such as zinc and copper (Marschner and Dell 1994). Furthermore, they can protect the plants against microorganisms, improve soil structure, and confer heavy metal resistance to plants (Leyval et al. 1997; Gonzalez-Chavez et al. 2002; Smith et al. 2003) and suppress aggressive agricultural weeds (Rinaudo et al. 2010).
While natural ecosystems have been estimated to contain up to 25–30 AMF species (Fitter 2001), levels of diversity of these organisms often decrease to two to seven species in arable lands, suggesting that a reduction in host plant diversity is paralleled with a similar reduction in the diversity of the mycobiota (Oehl et al. 2004; Li et al. 2007). Tchabi et al. (2008) found that AMF species richness decreased with land use intensification independent of the ecological zone. The decrease in diversity is attributed to the selective pressure of complex agricultural practices, such as plowing (Jansa et al. 2002, 2003; Roldan et al. 2007), fertilization (Bhadalung et al. 2005), and fungicide applications (Oehl et al. 2004), that strongly affect AMF communities. Besides these factors, soil type (Wang et al. 2008), soil depth (Yang et al. 2010), the abundance and/or composition of weeds (Salinas-Garcia et al. 2002; Oehl et al. 2004), and the previous crops also affect AMF population (Jefwa et al. 2006; Mathimaran et al. 2005; Vestberg et al. 2005). Although AM fungi have no host plant specificity, a number of studies demonstrated that plants, on the other hand, may prefer specific subgroups of these mycosymbionts (Oliveira et al. 2009).
Pioneer studies on the mycorrhizal communities associated with maize used conventional non-molecular approaches, including trap cultures, spore sieving, and morphological identification of spores. Based on such methods, Franke-Snyder et al. (2001) showed that the general structure of AM fungal communities was similar in conventional and low-input farming systems and management practices caused not much change in AMF diversity during 15 consecutive years of farming. Contrary to these findings, AMF diversity was found to be much lower in high-input continuous maize monoculture than in a moderate-input 7-year crop rotation system (Oehl et al. 2003). Long-term NP-fertilization reduced AMF diversity and decreased the total amount of AMF spores in maize rhizosphere, but arbuscular mycorrhiza species responded differentially to nutrient supply: The relative abundance of several Glomus species significantly decreased in response to fertilization, whereas other species, including Entrophospora schenckii, Glomus mosseae, and Scutellospora fulgida, were insensitive to fertilization (Bhadalung et al. 2005).
Molecular approaches are powerful tools for assessing the diversity of AM fungi (van Tuinen et al. 1998). By comparing sequence differences in small subunit ribosomal RNA genes, Daniell et al. (2001) demonstrated that AMF diversity was much lower in barley, maize, peas, and wheat than that found in a natural woodland site. Colonization of crops grown in frequently disturbed arable fields was dominated by two closely related Glomus sequence types indicating that intense soil cultivation reduces the diversity of AM fungi. Soil tillage practices were also found to affect AMF colonization of maize roots in a long-term field experiment run in Switzerland: PCR-based identification of AM fungus species showed that the occurrence of Scutellospora species was strongly reduced in plowed or chiseled soils, while fungi belonging to the suborder Glomineae were less sensitive to such practices (Jansa et al. 2003). The community composition of AM fungi in maize rhizosphere was greatly influenced by fertilizer amendments including calcium nitrate, ammonium sulfate, and organic manure in a long-term trial (Toljander et al. 2008). In these experiments, Glomus intraradices was the most frequently identified AM taxon, and it occurred in almost all treatments and was the only representative of AM fungi in nearly half of all the experimental plots. Maize genotypes may also influence the mycorrhizal community: AMF species richness was higher in the rhizosphere of phosphorous-efficient maize genotypes than that found in plots where phosphorous-inefficient genotypes were grown (Oliveira et al. 2009).
According to previous studies, maize monoculture, lasting for 5–28 years, reduce AMF diversity (Alguacil et al. 2008; Hijri et al. 2006; Oehl et al. 2003; Toljander et al. 2008). The present study takes the advantage of a 50-year-old continuous maize monoculture trial established at Martonvásár, Hungary with the aim to assess the effect of such an extreme reduction of host plant diversity on AMF community. An additional innovative aspect of the work was to examine some durably used agricultural measures on phylogenetic subgroups of Glomeraceae.
Materials and methods
Site description, agricultural practice, and sampling
A long-term maize monoculture experiment, established in 1958, located at Martonvásár (47°21′ N, 18°49′ E), Hungary was selected for sample collection. The climate of the region is classified as continental: The mean annual temperature and precipitation between 1958 and 2008 were 10.6°C and 539 mm, respectively. The soil of the experimental area was a humus loam of the chernozem type with forest residues (21%, 48%, and 18% clay, silt, and sand content, respectively), slightly acidic in the ploughed layer, with poor supplies of available phosphorus and good supplies of potassium. The experimental area was split to small plots (7 × 7 m plots for the different agrotechnical treatments and 14 × 7 m as non-treated control plots) that received the same treatment year after year: (a) mineral fertilization (MIN), started in 1967—200 kg nitrogen ha−1 as ammonium nitrate, 100 kg phosphorus ha−1 as P2O5, and 100 kg potassium ha−1 as K2O; (b) recycling crop residues (RCR), started in 1958—corn stalk residues either 5 t ha−1 (between 1958 and 1983) or 7.5 t ha−1 (since 1984 until now) were incorporated into the upper 25 cm of the soil by plowing; and (c) no fertilization, no soil cultivation (non-treated control, NON). The main physicochemical characteristics of the upper 25-cm soil layer of the differently managed plots are summarized in Table 1. Weeds and insects were controlled by pesticide treatments in all plots: Force (tephlutrine, 1.2 l ha−1), Acenit A 880 EC (acetochlore, 2.0 l ha−1), and a combination of Motivell (nicosulfuron) + Cambio (dicamba + bentason) + Dach HC surfactant (0.8 + 2.0 + 0.6 ha−1) have been used since 1995 as pre-sowing, pre-emergence, and post-emergence treatments, respectively.
Samples were collected at 13 June (start of flowering), 3 July (start of seed filling), 7 August (start of biological ripening), and 20 October (immediately after harvesting) in 2008. Three plants were dug out (with a soil core of ~30 × 30 × 30 cm) randomly from three repetitive plots of the same treatment. The total number of plant samples was 3 (individuals) × 3 (repetition) × 4 (sampling occasion) from treated (RCR, MIN) and 6 (individuals) × 3 (repetition) × 4 (sampling occasion) from control (NON) plots giving altogether 144 plants. The roots and the soil were stored in separate plastic bags in cool box (~4°C) until processing within 24 h.
Spore counts and assessment of root colonization
AMF spores were collected from 100 g air-dried soil samples using wet sieving (Gerdemann and Nicolson 1963) through 30 and 200 μm sieves followed by sucrose gradient centrifugation (Furlan et al. 1980). After centrifugation, spores and spore clusters were transferred into Petri dishes and counted using a stereomicroscope. Spore abundance in rhizosphere samples was expressed as the number of AMF spores per gram soil.
Roots were washed from soil cores, cut to 10 mm pieces, and three randomly selected pieces from each sample were subjected to Trypan Blue staining (Phillips and Hayman 1970). Internal fungal structures (hypae, arbuscules) were examined under a stereomicroscope at ×400 magnification, and the percentage of root length colonization was calculated using the gridline intersect method (Giovannetti and Mosse 1980).
Treatment effects on measured variables were subjected to variance analysis. Tukey’s honestly significantly difference was used as post hoc test. Data were analyzed using Statistica 6.1 (StatSoft, Tulsa, OK, USA) software.
Molecular analysis
Five randomly cut hairy root segments (~2–4 cm long) of each of the 72 plants collected in June and August (1st and 3rd sampling time) were subjected to DNA extraction using the DNeasy®plant Mini Kit (Quiagen, Chatsworth, CA, USA) following the manufacturer’s instructions. Fragments of 18S rDNA gene sequences were amplified by using the primer pairs AMV4.5F (5′-AAT TGG AGG GCA AGT CTG G-3′)–AMV4.5R (5′-AGC AGG TTA AGG TCT CGT TCG T-3′) and AMV4.5NF (5′-AAG CTC GTA GTT GAA TTT CG-3′)–AMV4.5NR (5′-CAC CCA TAG AAT CAA GAA AGA-3′) in the first and second nested PCR according to Saito et al. (2004). The PCR mixtures (20 μl) contained 2 μl 10× PCR buffer (Fermentas, Vilnius, Lithuania), 1.5 μl 25 mM MgCl2, 2 μl dNTP mix, 1.0 μl of each primer (40 mM), 1 μl of template DNA, 0.2 μl (5 U) Taq polymerase (Fermentas, Vilnius, Lithuania), and 11.3 μl milliQ water. For amplification, the following program was used: initial denaturation at 95°C for 10 min, followed by 20 (first PCR) and 40 cycles (second PCR) consisting of 30 s at 94°C, 30 s at 60°C, and 1 min at 72°C and a final extension of 72°C for 10 min. Amplification products obtained from the 360 purified DNA samples (five root segments × 72 plants) were separated by electrophoresis in 2% (w/v) agarose gels and stained with ethidium bromide. The PCR products were then purified by isolating the fragments of appropriate size (~650 bp) using the GFX PCR DNA and Gel Band Purification Kit (GE Healthcare, Amersham Biosciences, Amersham, UK). PCR products amplified from samples, collected at the same time, and from the same treatment were pooled as recommended by Renker et al. (2006), and these pools were cloned into pGEM®-T Easy vector (3,015 bp) by using the pGEM®-T Easy Vector System (Promega, Madison, WI, USA) and transformed into Escherichia coli DH5α. Plasmids were purified from 30 to 40 positive clones of each pool by using the Wizard® Plus SV Minipreps DNA Purification System Kit (Promega, Madison, WI, USA), and they were sent for sequencing to the IIT Biotech Bielefeld (Germany). The exact number of clones subjected to sequencing form a given pool depended on the frequency of clones with chimeric sequences found in that specific pool.
Sequence accession numbers
The partial rDNA gene sequences reported in this study have been deposited in the NCBI database under accession numbers GU598262 to GU598518.
Phylogenetic analysis
Sequence similarities were determined by using the BLASTn sequence similarity search program (Altschul et al. 1997) provided by the GenomNet website (http://www.genome.jp). Sequences blasted against the NCBI database (http://www.ncbi.nlm.nih.gov/) were aligned to reference sequences using the ClustalW 1.6 algorithm, version 1.6 (http://www.ebi.ac.uk/clustalw/), and they were converted into a distance matrix using DNADIST of PHYLIP. The program CHIMERA_CHECK 2.7 (Ribosomal Database Project II; http://rdp.cme.msu.edu) was used to select chimeric 18S DNA sequences. Chimeric sequences were omitted from further analysis. Rarefaction curves and diversity estimators (ACE, Chao) were calculated with the DOTUR program (Schloss and Handelsman 2005). DOTUR was also used to delineate operational taxonomic units (OTUs) at 97% similarity level. Sequences of representatives of these OTUs and 59 reference sequences from public databases were used to construct a phylogenetic tree by distance analysis with the program Molecular Evolutionary Genetics Analysis software 4.0 (Tamura et al. 2007) using neighbor-joining algorithms (Saitou and Nei 1987) and the Kimura two-parameter model. Bootstrap values were estimated from 1,000 replicates for the neighbor-joining analysis. The clones were grouped into several sequence types within the phylogenetic tree, and they were assigned to groups and subgroups according to the nomenclatural system used by Schwarzott et al. (2001) and Saito et al. (2004).
The Shannon–Wiener diversity index (H w) was calculated by using the DOTUR program from the equation: H w = −∑ p i (ln p i ), where p i is the proportion of individuals found in the i-th OTU. The sampling effort was assessed using ESTIMATES 7.5 (Colwell 2005) to randomize sample order in 100 replications.
Results
AMF spore abundance and mycorrhizal colonization of maize roots
These preliminary experiments were performed to decide the optimum sampling time to assess AM diversity by means of molecular analysis. Spore abundance was the lowest in samples taken in June, at the start of flowering when only ~3 spores g−1 were found in the rhizosphere soil. AMF total spore numbers increased with the progress of plant development reaching their maxima in August when 13–18 spores g−1 soil were found. A slow decrease in spore abundance was observed at harvesting in plots where normal mineral fertilization (MIN treatment) was applied or stalk residues were incorporated into the soil at 25 cm depth (RCR treatment). According to the Tukey test, mineral fertilization (MIN treatment) moderately but significantly (p = 0.0025) reduced the overall AMF spore abundance as compared to NON treatment (Fig. 1). The percentage of mycorrhizal root colonization assessed by Trypan Blue staining was not significantly different between samples collected from MIN, RCR, and non-fertilized control (NON) plots. The lowest degree of colonization of maize roots was observed at the start of flowering (in June). The colonization percentage increased to 50% by the start of biological ripening (in August), and then a slight decrease was found in all samples irrespective of fertilization or stalk residue incorporation (Fig. 2). Based on the high colonization activity observed at the start of biological ripening, samples collected in August promised the best insight into AMF diversity occurring in maize monoculture, and therefore, these samples were subjected to DNA extraction and PCR identification of fungi. Samples collected in June were also subjected such analysis with the aim to tag both the early and the mid-season colonizers.

The effects of sampling time and different agricultural practices on AMF spore abundance in the rhizosphere of maize grown in monoculture. Sampling times: June 13 June (1st), July 3 July (2nd), August 7 August (3rd), and October 20 October (4th). MIN plots received regular NPK fertilization, RCR corn stalk residues were incorporated into the upper 25 cm of the soil by plowing, NON no fertilization, no soil cultivation occurred. Each value is the mean of four replicates

Colonization of maize roots by AM fungi as affected by sampling times and agricultural practices. Sampling times: June 13 June (1st), July 3 July (2nd), August 7 August (3rd), and October 20 October (4th). MIN plots received regular NPK fertilization, RCR corn stalk residues were incorporated into the upper 25 cm of the soil by plowing, NON no fertilization, no soil cultivation occurred. Each value is the mean of six replicates
Phylogenetic analysis of PCR-amplified sequences
AM fungal sequences were found in all plants, i.e., in all three treatments and at both sampling times. Of the 257 sequences recovered from maize roots by PCR amplification, 203 (79%) belonged to Glomeromycota. The majority of the 203 Glomeromycota sequences belonged to the family Glomeraceae; altogether, 197 Glomeraceae sequences were identified, with a 194:3 distribution between groups Glomus A and Glomus B. Only one and five sequences were assigned to families Archaeosporaceae and Paraglomeraceae, respectively. From the 54 non-Glomeromycota sequences, three, 18, 15, and again 18 belonged to Blastocladiomycota, Basidiomycota, Chytridiomycota, and Zygomycota, respectively.
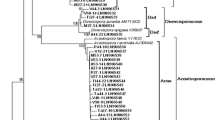
All Glomeromycota sequences identified in this study shared high similarity (94–100%) with sequences previously deposited at GenBank. By using the DOTUR program (Schloss and Handelsman 2005), these 203 Glomeromycota sequences were grouped into 22 OTUs at 97% similarity level. Representatives of these 22 OTUs and 59 reference sequences from public databases were used to construct a neighbor-joining tree (Fig. 3).

Phylogenetic tree displaying the relationship of sequences of AM fungi recovered from maize roots in the present study and 59 reference sequences. Bootstrap values are shown above the lines. Sequences obtained in this work are in bold and labeled with the phylogenetic subgroup symbol, the OTU number they were assigned to and the clone number. Reference sequences are labeled with the name provided by the database entry and the corresponding accession number. Phylogenetic group and subgroup assignments are shown on the right side of the tree (total number of sequences belonging to a specific OTU is given in Table 2)
Maize roots were mainly colonized by OTU 16, OTU 17, OTU 18, OTU 19, OTU 20, OTU 21, and OTU 22 (see Table 2). The cumulative number of clones belonging to these seven OTUs amounted 78.8% of the total number of Glomeromycota sequences identified in this study. OTU 22, the most abundant OTU, was closely related to Glomus sp. BEG126, while the next most frequent OTUs, OTU 20, and OTU 21 were very similar to G. intraradices and an unidentified Glomus sp. (GenBank accession number EU169397), respectively. Clones belonging to OTUs 18 and 19 clustered together with a yet undescribed taxon, Glomus sp. Glo3 (AJ715998).
Phylogenetic groups and subgroups were identified according to the classification proposed by Schwarzott et al. (2001) and Saito et al. (2004). The 194 unique sequences (18 OTUs) assigned to Glomus group A were evenly distributed among the four subgroups, Glomus Aa, Glomus Ab, Glomus Ac, and Glomus Ad. Glomus Ab comprising 70 unique sequences and Glomus Ac with 31 sequences were the most and the least densely represented subgroups, respectively.
The diversity of PCR-amplified AMF clones recovered from root samples collected from NON plots was higher than that of the clones from MIN and RCR plots, indicating that both mineral fertilization and crop residue recycling (accompanied with tillage-induced disruption of hyphae) reduce the diversity of AM fungi (Table 2). Clones belonging to subgroup Glomus Aa were absent in roots collected from MIN plots, but they were frequently detected in samples from the other two treatments (RCR, NON), suggesting that these AM fungi are sensitive to mineral fertilization but successfully tolerate soil disturbance. In general, clones belonging to subgroups Glomus Ab, Ac, and Ad rarely occurred in samples from RCR plots indicating the disturbance sensitivity of these phylotypes. Glomus Ad clones were not recovered from samples collected in MIN plots at the start of flowering, while these fungi actively colonized maize plants of similar age grown in NON plots. Glomus Ad fungi appeared again in maize roots grown in MIN plots, when the plants entered into the biological ripening stage.
The occurrence of other phylotypes was too low to draw any conclusion on factors supporting the colonization efficiency of these rare fungi. For example, the single Archaeosporaceae sequence was detected early in the colonization cycle in root samples from the MIN treatment, and the other two non-Glomeraceae OTUs, OTU 2 and OTU 13 represented by one and four clones, respectively, were also mainly observed in early growth stages.
A sampling effort curve (Fig. 4) was constructed to assess the influence of the sampling strategy on the AMF diversity found in samples collected from different agricultural treatments. For MIN plots, quite a flattened curve was obtained indicating that the number of OTUs we identified approached saturation and not much additional OTUs could be expected if the number of samples would have been considerably increased. AMF diversity found in NON and RCR plots was significantly higher than that detected in MIN plots, but again the curves for these plots showed a clear leveling off run, suggesting that the number of samples was enough to recover most potential OTUs that occurred in this long-term maize monoculture.

Sampling effort curve representing the relationships between OTU richness and the number of sequences for the three different agricultural practices. MIN plots received regular NPK fertilization, RCR corn stalk residues were incorporated into the upper 25 cm of the soil by plowing, NON no fertilization, no soil cultivation occurred
The Shannon–Wiener diversity index showed a clear difference between MIN/RCR plots and NON plots. AMF diversity found in roots from the NON plots (2.34 ± 0.16) was significantly higher than that observed in the other two treatments (RCR 1.79 ± 0.33, MIN 1.53 ± 0.21), and the number of OTUs recovered from RCR was also higher than that found in MIN, but the difference was statistically non-significant.
Figure 5 summarizes the effect of different agricultural treatments on phylogenetic subgroup composition of AM fungi. Cumulative numbers of clones amplified in the two sampling times, i.e., at the start of flowering (June) and biological ripening (August), respectively, were used to construct these diagrams. In samples from MIN plots, clones belonging to subgroup Glomus Ab were most frequently detected. The relative occurrence frequencies of Glomus Ac and Glomus Ad clones were nearly equal. Representatives of subgroup Glomus Aa were absent in plots where mineral fertilization was regularly applied. On the other hand, members of Glomus Aa dominated in samples from RCR plots. In samples from NON plots, clones belonging to the four subgroups of Glomus A were evenly distributed. Clones belonging to Archaeosporaceae, Paraglomeraceae, and Glomus group B occurred only sporadically.

Percentage distribution of AM fungi recovered from maize roots according to their phylogenetic assignment as affected by different agricultural treatments. MIN plots received regular NPK fertilization, RCR corn stalk residues were incorporated into the upper 25 cm of the soil by plowing, NON no fertilization, no soil cultivation occurred
Discussion
In agreement with previous results (Oehl et al. 2003, 2005; Hijri et al. 2006; Toljander et al. 2008), this study confirmed that a relatively high OTU richness can be found in arable lands, even in an extremely long-term maize monoculture. Sixteen OTUs were identified in maize roots grown in no-till, no fertilized plots (NON), nine OTUs in roots from plots where 200 kg N, 100 kg P, and 100 kg K ha−1 was applied yearly (MIN), and 13 OTUs were recovered from root samples collected in plots where stalk residues were incorporated in the soil every year (RCR). The present data confirmed earlier reports (Jansa et al. 2002; Roldan et al. 2007; Tchabi et al. 2008) on an inverse relationship between management intensity and AMF diversity but at the same time extended our knowledge on the behavior of AM fungi belonging to different phylogenetic subgroups. The Shannon–Wiener diversity values calculated from data of the present survey clearly demonstrated that both mineral fertilization and regular soil cultivation reduce diversity as compared to no-till, non-fertilized control, but these two agricultural measures have contrasting effects on phylogenetic subgroups within Glomus group A. Both treatments increased humus, phosphorus, and potassium contents of the soil (Table 1), and this might contribute to the reduction of AMF diversity observed in MIN and RCR plots.
Early studies based on spore trapping and morphological identification of spores might overestimate the diversity of AM fungi in maize stands as spore collection reveals rare species, detects symbiotically inactive fungi, and tags AM fungi colonizing weeds growing on the sampling area. On the other hand, PCR detection of fungi in root tissues, the approach we used in this study, identifies mainly the symbiotically active fungal structures, and therefore, these data more reliably reflect the community structure of AM fungi colonizing a plant species on target.
The 22 different OTUs discovered in this survey are certainly a minimum number of AMF phylotypes that occur in maize monoculture. However, the sampling effort curve we constructed from the randomized sample order strongly suggests that the majority of AM fungi that may occur in maize monoculture under the soil and climatic conditions prevailing in the experimental site were indeed recovered in this study. Plants, other than maize, had no influence on the AMF composition established at the experimental site sampled, as weeds were completely eradicated from all plots by regular herbicide treatments.
The vast majority of sequences identified in this assay belonged to Glomus group A. This finding was to be expected as other workers (Oehl et al. 2003; Hijri et al. 2006) have already shown that species of Archaeosporaceae and Paraglomeraceae, otherwise widely distributed in natural ecosystems, are rare components in AMF communities prevailing in arable systems. Omitting crop rotation may further reduce the diversity of AM fungi leading to an over-dominance of Glomeraceae. The occurrence of Archaeospora sp. in maize rhizosphere soil has been reported by Oliveira et al. (2009), but this type of AM fungi has never been identified previously in roots of maize. The single Archaeospora specimen identified in this survey was probably an active early colonizer of roots in NON plots, since inactive fragments or spores, present as contaminants on the root surface, were removed by intensive washing used prior to DNA isolation. In agreement with previous studies, AM fungi belonging to Paraglomeraceae were rarely found in our survey. Hijri et al. (2006) regularly found Paraglomus sp. in conventionally managed maize, but they could not recover these fungi from roots of maize grown in monoculture. According to Alguacil et al. (2008), members of Paraglomeraceae are rare components of the mycobiota of arable crops grown under warm, subtropical conditions. AM sequences clustered within Gigasporaceae have been identified in maize roots by PCR detection (Jansa et al. 2003; Alguacil et al. 2008), but these fungi are not frequently found in arable system. Gigasporaceae propagate by spore dispersal, their successful establishment requires an intact mycelial network (Daniell et al. 2001), and therefore, they have difficulties to survive in environments exposed to frequent physical disturbances caused by soil cultivation. Crop production in monoculture further reduces the establishment of these vulnerable AM fungi as neither previous workers (Hijri et al. 2006; Toljander et al. 2008), nor the present assessment could detect Gigaspora spp. in roots of maize grown in monoculture. The sporadic occurrence of clones belonging to Archaeosporaceae, Paraglomeraceae, and Glomus group B as confirmed in the present survey suggests that these AM fungi are not successful colonizers of maize grown in monoculture and even low-input farming (NON treatment) is unsuitable to increase their abundance.
Previous studies based on molecular identification of AM fungi showed the predominance of Glomus species in maize monoculture. Hijri et al. (2006) identified only three phylotypes all belonging to Glomus group A in roots from a continuous maize from a 28-year-old monoculture, whereas they could detect representatives of Paraglomus, Gigaspora, and Glomus group B only in roots collected from conventionally managed maize stands. On the other hand, AMF diversity within Glomus group A could be rather high as demonstrated by Toljander et al. (2008) who found a similar richness of taxonomical units within Glomus group A (Glomeraceae 1) in a 5-year-old maize monoculture as we found in this 50-year-old monoculture.
A number of studies (Daniell et al. 2001; Jansa et al. 2003) have explained the predominance of Glomus group A species in arable systems by the tolerance of these AM fungi to soil cultivation induced disruption of the mycelia network. In the present research, we observed significant differences within Glomus group A in response to different soil tillage practices. Representatives of Glomus Aa were the most frequently recorded specimens in plots where stalk residues were yearly incorporated in the soil, whereas members of Glomus Ab were rarely found in root samples collected from this treatment. On the other hand, Glomus Aa fungi were not detected in the mineral fertilization treatment, where they seemed to be replaced by AM fungi belonging to Glomus Ab.
In conclusion, the present study confirms and extends previous findings by Hijri et al. (2006) and Toljander et al. (2008) that Glomus group A fungi prevail in maize monoculture, but the AM diversity within this group can be rather high even under conditions where maize has been grown in exclusive monoculture for half a century. Furthermore, agricultural practices, including mineral fertilization and stalk rot incorporation, have a profound effect on diversity within Glomus group A and select for subgroups Glomus Ab and Glomus Aa, respectively. The future challenge is now to investigate the growth promoting efficiency of selected members of these two Glomus A subgroups in maize monoculture.
References
Alguacil MM, Lumini E, Roldan A, Salinas-Garcia JR, Bonfante P, Bianciotto V (2008) The impact of tillage practices on arbuscular mycorrhizal fungal diversity in subtropical crops. Ecol Appl 18:527–536. doi:10.1890/07-0521.1
Altschul SF, Madden TL, Schāffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucl Acids Res 25:3389–3402
Bhadalung NN, Suwanarit A, Dell B, Nopamornbodi O, Thamchaipenet A, Rungchuang (2005) Effects of long-term NP-fertilization on abundance and diversity of arbuscular mycorrhizal fungi under a maize cropping system. Plant Soil 270:371–382. doi:10.1007/s11104-004-1829-4
Colwell RK (2005) EstimateS: statistical estimation of species richness and shared species from samples. Version 8.0. User’s guide and application. Accessed 18 Jul 2008. Available: http://purl.oclc.org/estimates
Daniell TJ, Husband R, Fitter AH, Young JPW (2001) Molecular diversity of arbuscular mycorrhizal fungi colonising arable crops. FEMS Microbiol Ecol 36:203–209. doi:10.1111/j.1574-6941.2001.tb00841.x
Egner H, Riehm H, Mingo WR (1960) Untersuchungen über die chemische Bodenanalyse als Grundlage für die Beurteilung des Nährstoffzustandes der Böden. Kungl Lantbrukshögsk Ann Uppsala 26:199–215
Fitter AH (2001) Specificity, links and networks in the control of diversity in plant and microbial communities. In: Huntly MC, Levin S (eds) Ecology: achievement and challenge. British Ecological Society symposium. Blackwell Scientific, Oxford, pp 95–114
Franke-Snyder M, Douds DD, Galvez L, Phillips JG, Wagoner P, Drinkwater L, Morton JB (2001) Diversity of communities of arbuscular mycorrhizal (AM) fungi present in conventional versus low-input agricultural sites in eastern Pennsylvania, USA. Appl Soil Ecol 16:35–48. doi:10.1016/S0929-1393(00)00100-1
Furlan V, Bartschii H, Fortin JA (1980) Media for density gradient extraction of endomycorrhizal spores. Trans Brit Mycol Soc 75:336–338. doi:10.1016/S0007-1536(80)80102-1
Gerdemann JW, Nicolson TH (1963) Spore of mycorrhizal endogene species extracted from soil by wet sieving and decanting. Trans Brit Mycol Soc 46:235–244. doi:10.1016/S0007-1536(63)80079-0
Giovannetti M, Mosse B (1980) An evaluation of techniques to measure vesicular–arbuscular infection in roots. New Phytol 2:489–500. doi:10.1111/j.1469-8137.1980.tb04556.x
Gonzalez-Chavez C, D’haen J, Vangronsveld JJ, Dodd JC (2002) Copper sorption and accumulation by the extraradical mycelium of different Glomus spp. (arbuscular mycorrhizal fungi) isolated from the same polluted soil. Plant Soil 240:287–297. doi:10.1023/A:1015794622592
Hijri I, Sýkorová Z, Oehl F, Ineichen K, Mäder P, Wiemken A, Redecker D (2006) Communities of arbuscular mycorrhizal fungi in arable soils are not necessarily low in diversity. Mol Ecol 15:277–289. doi:10.1111/j.1365-294X.2006.02921.x
Jansa J, Mozafar A, Anken T, Ruh R, Sanders IR, Frossard E (2002) Diversity and structure of AM communities as affected by tillage in a temperate soil. Mycorrhiza 12:225–234. doi:10.1007/s00572-002-0163-z
Jansa J, Mozafar A, Kuhn G, Anken T, Ruh R, Sanders IR, Frossard E (2003) Soil tillage affects the community structures of mycorrhizal fungi in maize roots. Ecol Appl 13:1164–1176. doi:10.1890/1051-0761(2003)13[1164:STATCS]2.0.CO;2
Jefwa JM, Sinclair R, Maghembe JA (2006) Diversity of glomale mycorrhizal fungi in maize/sesbania intercrops and maize monocrop systems in southern Malawi. Agrofor Syst 67:107–114. doi:10.1007/s10457-004-2370-4
Leyval C, Turnau K, Haselwandter K (1997) Effect of heavy metal pollution on mycorrhizal colonization and function: physiological, ecological and applied aspects. Mycorrhiza 7:139–153. doi:10.1007/s005720050174
Li LF, Li T, Zhao ZW (2007) Differences of arbuscular mycorrhizal fungal diversity and community between a cultivated land, an old field, and a never-cultivated field in a hot and arid ecosystem of southwest China. Mycorrhiza 17:655–665. doi:10.1007/s00572-007-0143-4
Maherali H, Klironomos JN (2007) Influence of phylogeny on fungal community assembly and ecosystem functioning science. Science 316:1746–1748. doi:10.1126/science.1143082
Marschner H, Dell B (1994) Nutrient uptake in mycorrhizal symbiosis. Plant Soil 159:89–102. doi:10.1007/BF00000098
Mathimaran N, Ruh R, Vullioud P, Frossard E, Jansa J (2005) Glomus intraradices dominates arbuscular mycorrhizal communities in a heavy textured agricultural soil. Mycorrhiza 16:61–66. doi:10.1007/s00572-005-0014-9
Oehl F, Sieverding E, Ineichen K, Mader P, Boller T, Wiemken A (2003) Impact of land use intensity on the species diversity of arbuscular mycorrhizal fungi in agroecosystems of central Europe. Appl Environ Microbiol 69:2816–2824. doi:10.1128/AEM.69.5.2816-2824.2003
Oehl F, Sieverding E, Ineichen K, Mäder P, Dubois D, Boller T, Wiemken A (2004) Impact of long-term conventional and organic farming on the diversity of arbuscular mycorrhizal fungi. Oecologia 138:574–583. doi:10.1007/s00442-003-1458-2
Oehl F, Sieverding E, Ineichen K, Ris EA, Boller T, Wiemken A (2005) Community structure of arbuscular mycorrhizal fungi at different soil depths in extensively and intensively managed agroecosystems. New Phytol 165:273–283. doi:10.1111/j.1469-8137.2004.01235.x
Oliveira CA, Sá NMH, Gomes EA, Marriell IE, Scotti MR, Guimaraes CT, Schaffert RE, Alves VMC (2009) Assessment of the mycorrhizal community in the rhizosphere of maize (Zea mays L.) genotypes contrasting for phosphorus efficiency in the acid savannas of Brazil using denaturing gradient gel electrophoresis (DGGE). Appl Soil Ecol 41:249–258. doi:10.1016/j.apsoil.2008.11.005
Phillips JM, Hayman DS (1970) Improved procedures for clearing roots and staining parasitic and vesicular–arbuscular mycorrhizal fungi for rapid assessment of infection. Trans Brit Mycol Soc 55:158–161
Renker C, Weißhuhn K, Kellner H, Buscot F (2006) Rationalizing molecular analysis of field-collected roots for assessing diversity of arbuscular mycorrhizal fungi: to pool, or not to pool, that is the question. Mycorrhiza 16:525–531. doi:10.1007/s00572-006-0067-4
Rinaudo V, Bàrberi P, Giovannetti M, van der Heijden MG (2010) Mycorrhizal fungi suppress aggressive agricultural weeds. Plant Soil 333:7–20. doi:10.1007/s11104-009-0202-z
Roldan A, Salinas-Garcia JR, Alguacil MM, Caravaca F (2007) Soil sustainability indicators following conservation tillage practices under subtropical maize and bean crops. Soil Till Res 93:273–282. doi:10.1016/j.still.2006.05.001
Saito K, Suyama Y, Sato S, Sugawara K (2004) Defoliation effects on the community structure of arbuscular mycorrhizal fungi based on 18S rDNA sequences. Mycorrhiza 14:363–373. doi:10.1007/s00572-003-0286-x
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Salinas-Garcia JR, Velazquez-Garcia JJ, Gallardo-Valdez M, Díaz-Mederos P, Caballero-Hernández F, Tapia-Vargas LM, Rosales-Robles E (2002) Tillage effects on microbial biomass and nutrient distribution in soils under rain-fed corn production in central-western Mexico. Soil Till Res 66:143–152. doi:10.1016/S0167-1987(02)00022-3
Schloss PD, Handelsman J (2005) Introducing DOTUR a computer program for defining operational taxonomic units and estimating species richness. Appl Environ Microbiol 71:1501–1506. doi:10.1128/AEM.71.3.1501-1506.2005
Schwarzott D, Walker C, Schüßler A (2001) Glomus, the largest genus of the arbuscular mycorrhizal fungi (Glomales), is nonmonophyletic. Mol Phylogenet Evol 21:190–197. doi:10.1006/mpev.2001.1007
Smith SE, Smith FA, Jakobsen I (2003) Mycorrhizal fungi can dominate phosphate supply to plants irrespective of growth responses. Plant Physiol 133:16–20. doi:10.1104/pp.103.024380
Sylvia DE, Hammond LC, Bennet JM, Hass JH, Linda SB (1993) Field response of maize to a VAM fungus and water management. Agron J 85:193–198
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599. doi:10.1093/molbev/msm092
Tchabi A, Coyne D, Hountondji F, Lawouin L, Wiemken A, Oehl F (2008) Arbuscular mycorrhizal fungal communities in sub-Saharan savannas of Benin, West Africa, as affected by agricultural land use intensity and ecological zone. Mycorrhiza 18:181–195. doi:10.1007/s00572-008-0171-8
Theuri S, Buscot F (2010) Laccases: toward disentangling their diversity and functions in relation to soil organic matter cycling. Biol Fertil Soils 46:215–226. doi:10.1007/s00374-010-0440-5
Toljander JF, Santos-González JC, Tehler A, Finlay RD (2008) Community analysis of arbuscular mycorrhizal fungi and bacteria in the maize mycorrhizosphere in a long-term fertilization trial. FEMS Microbiol Ecol 65:323–338. doi:10.1111/j.1574-6941.2008.00512.x
van der Heijden M, Klironomos JN, Ursic M, Moutoglis P, Streitwolf-engel R, Boller T, Wiemken A, Sanders IR (1998) Mycorrhizal fungal diversity determines plant biodiversity, ecosystem variability and productivity. Nature 396:69–72. doi:10.1038/23932
van Tuinen D, Jacquot E, Zhao B, Gollotte A, Gianinazzi-Pearson V (1998) Characterization of root colonization profiles by a microcosm community of arbuscular mycorrhizal fungi using 25S rDNA-targeted nested PCR. Mol Ecol 7:879–887. doi:10.1046/j.1365-294x.1998.00410.x
Vestberg M, Saari K, Kukkonen S, Hurme T (2005) Mycotrophy of crops in rotation and soil amendment with peat influence the abundance and effectiveness of indigenous arbuscular mycorrhizal fungi in field soil. Mycorrhiza 15:447–458. doi:10.1007/s00572-005-0349-2
Wang YY, Vestberg M, Walker C, Zhang XP, Lindström K (2008) Diversity and infectivity of arbuscular mycorrhizal fungi in agricultural soils of the Sichuan Province of mainland China. Mycorrhiza 18:59–68. doi:10.1007/s00572-008-0161-x
Yang FY, Li GZ, Zhang DE, Christie P, Li XL, Gai JP (2010) Geographical and plant genotype on the formation of arbuscular mycorrhizae in Avena sativa and Avena nuda at different soil depths. Biol Fertil Soils 46:435–445. doi:10.1007/s00374-010-0450-3
Acknowledgments
The authors thank Dr. Zoltán Berzsenyi for valuable advice and helpful comments on field experiment. This study was supported by grants from the National Research Council (OTKA K 76067) and by the National Office for Research and Technology (OM-00096, OMFB-0046/2009). L. H. thanks the support from the Office for Subsidized Research Units of the Hungarian Academy of Sciences.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sasvári, Z., Hornok, L. & Posta, K. The community structure of arbuscular mycorrhizal fungi in roots of maize grown in a 50-year monoculture. Biol Fertil Soils 47, 167–176 (2011). https://doi.org/10.1007/s00374-010-0519-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00374-010-0519-z