
Abstract
The plant hormone group, the cytokinins, regulates many stages of plant growth and development. Regulation includes that of cell division and enhancement of sink strength, both of which are important processes in seed development and embryonic growth. Two gene families play a key role in maintaining cytokinin homeostasis: isopentenyl transferase (IPT), which catalyzes the rate-limiting step in the formation of cytokinins, and cytokinin oxidase/dehydrogenase (CKX), which irreversibly inactivates cytokinins by cleaving the N6 side chain. Quantitative reverse transcriptase polymerase chain reaction (PCR) was used to measure the expression of individual gene family members to investigate the source of cytokinin and its subsequent inactivation during the early stages of seed and pod development. In this study, rapid-cycling Brassica rapa (RCBr) was used because of its genetic relatedness to commercial Brassica species, its rapid life cycle, its small adult size, and its larger reproductive organs compared to Arabidopsis. Our results indicate that BrIPT1, -3, and -5 and BrCKX1, -2, -3, and -5 express differentially both temporally and spatially within RCBr root, stem, leaf, seed, and pod tissues. Particularly strong expression was shown by IPT3 and IPT5 and CKX2 in developing seeds.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
From the time endogenous cytokinins were first identified in plants, these hormones have been shown to accumulate during the phase of cell division in developing fruit and seeds (Letham 1963). The origin of this cytokinin was initially proposed to be from the roots (see Noodén and Leopold 1978). However, it was recognized in early metabolism studies that root-supplied cytokinin does not provide sufficient cytokinin for seed development. In studies in which radiolabeled cytokinin was supplied to the xylem stream of leguminous plants, intact cytokinin moieties were detected in the pod and low levels in the seed coat, but intact cytokinin moieties were not detected in the embryo (for example, Jameson and others 1987; Singh and others 1988), even though labeled adenosine supplied in a similar manner was detected reaching the seed (Noodén and Letham 1984).
Since these early studies, it has been recognized that the rate-limiting step in cytokinin biosynthesis is controlled by isopentenyl transferase (IPT) (Kakimoto 2001) and, further, that this enzyme is coded for by a gene family with members expressed specifically in different tissues and/or developmental stages (Miyawaki and others 2004). Likewise, one of the key metabolism enzymes, cytokinin oxidase/dehydrogenase (CKX), also exists as a gene family (Schmülling and others 2003) and with individual specificities in terms of tissues and developmental stage (Vyroubalova and others 2009).
In Zea mays, the temporal and spatial expression and functional analysis of a specific IPT, ZmIPT2, is confirmation that maize seed biosynthesizes its own endogenous cytokinin (Brugière and others 2008). Previously, Brugière and others (2003) showed that expression of ZmCKX1 also occurred during early seed development. Brugière and others (2008) suggested that ZmIPT2 and ZmCKX1 are the gene family members controlling cytokinin homeostasis in the developing maize grain. In rice, Ashikari and others (2005) showed conclusively that OsCKX2 has a controlling role in grain production. A quantitative trait locus (QTL) associated with increased grain productivity in fact was a mutated cytokinin oxidase gene. Most recently, RNAi was used in barley to silence HvCKX1, with a resultant increase in both grain number and grain weight (Zalewski and others 2010).
However, although there are a number of examples in the literature that indicate that regions of high IPT expression or high endogenous cytokinin are matched by similar CKX activity (Galis and others 2005), simultaneous monitoring of IPT and CKX gene family members has only recently been reported (Vyroubalova and others 2009). Of relevance to this study is the specific upregulation of ZmIPT2, ZmCKX1, and ZmCKX4 gene family members in early-developing kernels of maize. Such information is lacking for pods and seeds of eudicots, including brassicas. Detailed information on the level of the gene family member is required if modern methods of plant breeding, such as TILLING, are to be utilized.
Rapid-cycling brassicas have been bred for use in genetic studies and can be used as an alternative model plant to Arabidopsis thaliana. Initially developed as a model for probing the genetic basis of plant disease (Musgrave 2000), RCBr develops rapidly and has both small adult size and a brief life cycle. Under optimal laboratory conditions, RCBr flowers within 16 days of seed germination and has a life cycle of 35–40 days, from parental seed sown to offspring seed harvest (Williams and Hill 1986). Because of their close relationship with the economically important Brassica species, rapid-cycling Brassica populations, especially those of B. rapa and B. oleracea, have seen widespread application in plant and crop physiology investigations. Another benefit of using RCBr is that the reproductive organs are larger than the model plant A. thaliana. The flowers of RCBr are three times larger than flowers from A. thaliana (Weinig cited by Kelly 2006) and seeds and pods are similar in size to those of forage brassica. Musgrave (2000) identified RCBr as a useful model for the investigation and improvement of seed storage reserves in Brassica. With this in mind, we selected RCBr as the model plant in this study.
In this article we report the identification and expression, using quantitative reverse transcriptase polymerase chain reaction (qRT-PCR), of several IPT and CKX gene family members during the early stages of pod and seed development in rapid-cycling Brassica rapa (RCBr).
Materials and Methods
Plant Material
Seeds of RCBr were obtained from Wisconsin Fast Plants (www.fastplants.org) and grown in trays of 60 pottles, in general potting mix, in a controlled growth room set at 25°C. Plants were supplied with 24-h lighting (PPFD ~ 300 μmol m−2 s−1) and continuous water supply as per the supplier’s instructions. Starting 12 days after germination, flowers were hand-pollinated and labeled, continuing until 17 days after germination. Samples were taken at selected stages of plant development. The stages sampled included 1–2-day-old leaves taken from around the developing flowers, fully expanded leaves harvested when the pods were starting to develop, and mature leaves taken from near the base of the plant when the pods were fully formed; young flower buds, flower buds with petals just emerging, and fully opened flowers (Stages 1, 2, and 3, respectively; Fig. 1); and four stages of pod and seed development [1, 2, 7, and 14 days after pollination (DAP)]. Root tips and stems from a mature plant were also harvested. Two independent experiments were run: From the first experiment, whole pods were extracted starting at 1 DAP as well as leaves, stems, and roots; from the second experiment, seeds were separated from pods starting from 4 DAP and analyzed independently.

The three stages of flowers used for analysis of gene expression. 1: Young flower buds. 2: Yellow petals just emerging from the buds. 3: Flowers are fully opened and able to be pollinated
RNA Extraction and cDNA Synthesis
Tissue samples were placed briefly in liquid nitrogen immediately upon harvest and stored at −80°C until required. Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following the manufacturer’s protocol. The integrity of the total RNA was determined by electrophoresis on 1% (w/v) agarose gel. The concentration and purity of the total RNA extracted was determined using a Nanodrop spectrophotometer (Nanodrop Technologies Inc., Wilmington, DE, USA) at the 260/280-nm ratio with expected values between 1.7 and 2.1.
For cDNA synthesis, l μg total RNA, 50 U Expand Reverse Transcriptase (F. Hoffman-La Roche Ltd, Basel, Switzerland), and 50 pmol oligo(dT) primers were used in a 20-μl reverse transcription reaction. The final reaction mix was incubated at 42°C for 2 h. The cDNA was diluted 1:10 with water.
Gene Isolation and Sequence Analysis
For degenerate primer design, A. thaliana IPT and CKX gene family members were used as templates to search the NCBI database for homologous B. rapa nucleotide and expressed sequence tag (EST) sequences. Retrieved sequences were then aligned using ClustalX 1.8. PCR primers for each family member were designed within the regions that are conserved across the reference sequences from the same family member but specific against other family members, using Primer Premier 5.0 (Supplementary Table 1). PCR amplification was conducted in a 20-μl reaction containing 2 mM MgCl2, 10 pmol of each forward and reverse primer, 2 μl of cDNA, 1 U Taq polymerase. After initial denaturation at 95°C for 5 min, the reaction was incubated at 94°C for 1 min, 52–55°C for 1 min (dependent on optimum PCR efficiency of primer pairs), and 72°C for 1 min, cycled 36 times, with a final elongation step at 72°C for 5 min, for amplification of the PCR product. The PCR products were separated on a 1% agarose gel.
Bands of the expected size were excised under blue light. The PCR product was extracted and purified using the Agarose Gel DNA Extraction Kit (Roche, Ref. 11 696 505 001). The amount of PCR product was quantified on an agarose gel using both a low DNA mass ladder (Roche) and a NanoDrop spectrophotometer. The purified PCR product was directly sequenced in an ABI 3700 sequencer using the Big Dye Terminator Reaction v3.0 (Applied Biosystems, Foster City, CA, USA). Independent sequences of both DNA strands were obtained using the same primers as used for RT-PCR. The sequences were then used to BLAST search the NCBI database to verify the nature of the putative IPT or CKX homologs.
qRT-PCR
qRT-PCR was carried out essentially as described by Song and others (2008). The geometric means of the housekeeping genes β-actin and GAPDH were used to normalize expression. The qRT-PCR reactions were performed in a 96-well Thermocycler (Stratagene, La Jolla, CA, USA) with SYBR® Green reaction mix (produced in-house), for 10 min at 95°C, followed by 40 runs of 45 s at 95°C, 45 s at 55°C, and 45 s at 72°C. The results were obtained using two biological replicates and three technical replicates. To account for any differences between individual runs, each plate run was either all IPT or all CKX samples and one each of actin and GAPDH samples. The error bars shown in Figs. 3 and 4 are 2 to the power of the standard deviation of the CT values (corrected relative to the housekeeping genes).
Chlorophyll Assays
Leaf tissue was ground, followed by the immediate addition of N,N-dimethylformamide and incubated in the dark at 4°C overnight. The following day the tubes were briefly centrifuged to pellet the ground leaf tissue. Three samples were individually analyzed and absorbances (Abs) read at 664 and 647 nm. The chlorophyll concentrations were calculated using the equations of Porra (2002).
Results
From the sequences obtained from the degenerate primers, BLAST searches were used to confirm homology to the Arabidopsis sequences originally used as templates. Phylogenetic trees of both IPT (Fig. 2a) and CKX (Fig. 2b) gene family members were produced. Each tree was rooted to Rhodococcus fascians homologs with previously identified gene family members from several species of plants (both monocotyledons and dicotyledons) to compare the relationship of the isolated genes to their homologs. The trees show that all of the newly isolated genes in the present study were most similar to their Arabidopsis homologs.

Phylogenetic tree of IPT family members (a) and CKX family members (b) rooted to Rhodococcus fascians. Representative sequences were obtained from NCBI databases. Sequences isolated in this study are in bold. Numbers at the nodes indicate the bootstrap values (%) ≥50. Products sequenced from the primers designed for this experiment (Supplementary Table 1) are in bold. Generic and species names as follows: A. thaliana, Brassica oleracea, B. rapa, Dendrobium huoshanense, Gossypium hirsutum, Hordeum vulgare, Oryza sativa, Populus trichocarpa, Ricinus communis, R. fascians, Solanum tuberosum, Zea mays
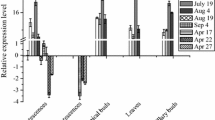
Differential expression of IPT and CKX gene family members is apparent both temporally and spatially. Expression levels of IPT1, -3, and -5 varied considerably across the tissue samples (Fig. 3a). There was over 110-fold more IPT1 mRNA in the highest-expressing tissues compared to the lowest. IPT1 was expressed during flower development, but was at a low level at the time the mature flowers were ready to be pollinated. Expression of IPT1 in the pods was minimal just after pollination but increased up to 7 DAP. In contrast to IPT1, expression of IPT3 and IPT5 increased only once pollination had occurred.

RT-qPCR gene expression of a IPT gene family members, b CKX gene family members from buds and flowers (flowers 1, 2, and 3, depicting bud to fully open flower); pods from 1-, 2-, 7-, and 14-DAP; leaves (leaves 1, 2, and 3, depicting youngest to oldest); root tips and stems. Bars show +SD of the mean of two biological and three technical replicates based on corrected CT number
In the second experiment (Fig. 4a), it is clear that the majority of IPT1, -3, and -5 expression occurred in the seeds rather than in the pods and that this is mostly the consequence of the expression of IPT3 and IPT5. Expression of all three genes was significantly less in the pods, with IPT5 showing expression specific to the seeds and no expression at all in the pods 14 DAP.

RT-qPCR gene expression of a IPT gene family members, b CKX gene family members extracted from pod cases and whole seeds, 4, 7, 11, and 14 DAP. Bars show +SD of the mean of two biological and three technical replicates based on corrected CT number
CKX5 expression was slightly elevated in developing flowers but decreased as the flowers matured (Fig. 3b), whereas CKX2 showed some activity in the mature flowers. Expression of CKX1, -2, -3, and -5 was low in pods immediately after pollination and highest in the tissue 7 DAP, with expression of CKX2 significantly higher than other gene family members. In the second experiment, when the pods and seeds were separated (Fig. 4b), CKX2 was expressed very highly in seeds relative to pods and other CKX family members, particularly at 14 DAP.
In leaves, expression of IPT1, -3, and -5 was lowest in the expanded leaves relative to the youngest and oldest leaves. The most significant activity is that of IPT3, which was very highly expressed in the mature leaves. A similar pattern of activity was seen for CKX1, -2, -3, and -5, where the expression of these gene family members decreased as the leaves expanded and then increased as the leaves matured. Expression of IPT1, -3, and especially -5 in the roots was higher than in the stem. For CKX2, -3, and -5, the opposite was the case where their expression was somewhat, though not markedly, higher in the stem compared to the roots, although expression of CKX1 was higher in the roots compared to the stems.
Discussion
In plants, the rate-limiting enzyme in cytokinin biosynthesis is considered to be IPT, which functions by attaching the isopentenyl side chain to the N6 moiety of ADP or ATP. The Arabidopsis genome has nine IPT gene family members (Kakimoto 2001; Takei and others 2001). Because AtIPT2 and AtIPT9 are tRNA isopentenyl transferases (Miyawaki and others 2006), they were not investigated. To date we have isolated three IPT homologs, IPT1, -3, and -5, from Brassica rapa. Homology to existing Arabidopsis, Brassica, and other previously identified genes is shown in the phylogenetic tree (Fig. 2a). The tree was rooted to R. fascians to enable an easier direct comparison to other IPT family members. IPT1, -3, and -5 cluster with their related brassica homologs and not with the more distantly related monocots. There are no Brassica rapa ESTs or nucleotide sequences in publicly available databases that show any homology to AtIPT6 or AtIPT4 and, apart from one 200-nucleotide sequence, no other sequence data are available for a homolog of AtIPT8. Kakimoto (2001) found that in some cultivars of Arabidopsis and rice, AtIPT6 and OsIPT6, respectively, appear to be pseudogenes whereby a nucleotide deletion caused a frame shift. Our attempts to isolate an IPT8 homolog have yet to be successful. Miyawaki and others (2004) showed that both AtIPT4::GUS and AtIPT8::GUS are expressed specifically in the chalazal region of the endosperm of Arabidopsis seeds. In a transcriptome analysis of Arabidopsis endosperm, using laser capture microdissection, Day and others (2008) detected AtIPT8 as well as numerous genes associated with cytokinin signaling.
It is clear that both BrIPT3 and BrIPT5 are highly expressed in seeds and BrIPT3 is also expressed in the oldest leaves sampled. In Arabidopsis, AtIPT5 was found to be expressed in lateral root primordia, columella root caps, upper parts of young inflorescences, and fruit abscission zones, whereas AtIPT3 was expressed in phloem companion cells (Takei and others 2004; Sakakibara 2006). Such differences between Arabidopsis and RCBr could be pursued by promoter::GUS reporter gene studies, which would allow identification of the specific cell groups in which the RCBr genes are expressed.
BrIPT1, -3, and -5 were not highly expressed in young developing pods, suggesting that maternally supplied cytokinin may be implicated in early pod development. The expression of BrIPT3 in the oldest leaves is interesting because cytokinin is normally associated with delay of senescence and unloading from the phloem into sink tissues (Lara and others 2004). As the pods and seeds are developing, these leaves would be expected to function as source leaves, so this high level of activity of one IPT gene family member warrants further investigation. Increased longevity of leaves is considered a target for increasing seed yield (Ma and others 2008).
Metabolism of cytokinins is complex and involves irreversible deactivation by CKX as well as a complex array of glucose conjugations, which, in the case of at least O-glucosylation of the side chain, are reversible. Moreover, the form of metabolism (deactivation or conjugation) has been shown to be both species- and tissue-specific (Jameson 1994). That CKX has a critical role in seed development is clear from work in which CKX is either over- or underexpressed in developing seeds. For example, when Kopečný and others (2006) overexpressed a CKX from maize in Arabidopsis, the plants displayed a phenotype typical of cytokinin deficiency, including reduced shoot apical meristem size and cell number, a reduced number of stamens, and, critically, shorter siliques that contained aborted seeds or seeds maturing abnormally. Earlier, Werner and others (2003) had shown similar results when AtCKX1 and AtCKX3 were overexpressed in Arabidopsis, but they also noted that of those seeds that did mature, both viable seeds and embryos were enlarged. In GUS reporter gene studies, CKX6::GUS was shown to express in the gynaecium and in the funiculus of the developing seeds.
The importance of CKX was shown conclusively by Ashikari and others (2005), whose study of rice found that plants that were null OsCKX2 mutants produced up to 24% more grain per panicle than commonly grown strains. Most recently, in yet another cereal, Zalewski and others (2010) showed that downregulation of barley HvCKX1 using RNAi technology resulted in an increase in yield through increases in both grain number and grain weight.
Because we are interested in gene targets for increasing seed yield in brassica species and because a reduction in the activity of CKX has been shown to lead to increased seed yield in cereals, we focused on CKX rather than the glucosylation enzymes. The Arabidopsis genome contains seven CKX gene family members (Bilyeu and others 2001). Cytokinin oxidase/dehydrogenase enzymes inactivate cytokinins irreversibly in a single enzymatic step by cleaving the N6 side chain from the adenine/adenosine moiety, converting active cytokinins such as zeatin and iP to adenine (Morris and others 1999; Galuska and others 2001; Massonneau and others 2004). To date, we have isolated four putative CKX homologs: BrCKX1, -2, -3, and -5 from Brassica rapa. Homology to existing Arabidopsis, Brassica, and other previously identified genes is shown in the phylogenetic tree (Fig. 2b). Not surprisingly, BrCKX1 aligns most closely to other brassica CKX1 and to AtCKX1; BrCKX2, -3, and -5 align most closely to AtCKX2, AtCKX3, and AtCKX5, respectively.
Similar to the key role shown for CKX gene family members in rice (Ashikari and others 2005), maize (Brugière and others 2003), and barley (Zalewski and others 2010), we now show in a eudicot that the expression of BrCKX2 from B. rapa, which increased substantially as pods matured, was associated with the developing seeds rather than the pod cases.
When IPT and CKX expressions are compared, there is a noticeable trend in all tissues studied: As IPT expression increases and decreases, CKX expression mimics this pattern. There are suggestions in the literature that this is a causative relationship (for example, Jones and Schreiber 1997; Galis and others 2005), and indeed Motyka and others (1996) showed that cytokinin oxidase activity was increased in transgenic ipt tobacco callus. However, Vyroubalova and others (2009) indicate that although most CKX gene family members responded to exogenous cytokinin by an increase in expression, ZmCKX3 was in fact downregulated.
Relatively higher expression of BrIPT1, -3, and particularly -5 in roots compared to stem tissue was not unexpected considering the long acceptance that root-produced cytokinins translocate to aboveground organs (Kudo and others 2010). Miyawaki and others (2004) also showed AtIPT5 to be upregulated in roots. We suggest root- and/or leaf-supplied cytokinin may be impacting pod (but not seed) development.
Conclusions
The RT-qPCR data presented here involved simultaneous monitoring of three members of the BrIPT gene family and four members of the BrCKX gene family. The data presented support suggestions that CKX plays a major role in maintaining cytokinin homeostasis during seed development and that downregulation of CKX, specifically in developing seeds, would appear to be a logical next target for improvement of seed storage reserves in brassica species. The data also support early metabolism studies that point to the necessity for seed-based biosynthesis of cytokinins, but possibly some dependence of the maternal tissue on cytokinin supplied from elsewhere in the plant because there is minimal expression, at least of BrIPT1, -3, and 5, at the very early stages of pod development.
References
Ashikari A, Sakakibara H, Lin S, Yamamoto T, Takashi T, Nishimura A, Angeles AR, Qian Q, Kitano H, Matsuoka M (2005) Cytokinin oxidase regulates rice grain production. Science 309:741–745
Bilyeu KD, Cole JL, Esparza TJ, Kramer MD, Laskey JG, Riekhof WR, Morris RO (2001) Molecular and biochemical characterization of a cytokinin oxidase from maize. Plant Physiol 125:378–386
Brugière N, Shuping J, Hanke S, Zinselmeier C, Roessler JA, Niu X, Jones RJ, Habben JE (2003) Cytokinin oxidase gene expression in maize is localized to the vasculature, and is induced by cytokinins, abscisic acid, and abiotic stress. Plant Physiol 132:1228–1240
Brugière N, Bohn J, Humbert S, Rizzo N, Habben J (2008) A member of the maize isopentenyl transferase gene family, Zea mays isopentenyl transferase 2 (ZmIPT2), encodes a cytokinin biosynthetic enzyme expressed during kernel development. Plant Mol Biol 67:215–229
Day RC, Herridge R, Ambrose BA, Macknight RC (2008) Transcriptome analysis of proliferating Arabidopsis endosperm reveals biological implications for the control of syncytial division, cytokinin signaling, and gene expression regulation. Plant Physiol 148:1964–1984
Galis I, Bilyeu KD, Godinho MJG, Jameson PE (2005) Expression of three Arabidopsis cytokinin oxidase/dehydrogense promoter:GUS chimeric constructs in tobacco: response to developmental and biotic factors. Plant Growth Regul 45:173–182
Galuska P, Frébort I, Sebela M, Sauer P, Jacobsen S, Pec P (2001) Cytokinin oxidase or dehydrogenase? Mechanism of cytokinin degradation in cereals. Eur J Biochem 268:450–461
Jameson PE (1994) Cytokinin metabolism and compartmentation. In: Mok DWS, Mok MC (eds) Cytokinins: chemistry, activity, and function. CRC Press, Boca Raton, FL, pp 113–124
Jameson PE, Badenoch-Jones J, Letham DS, Parker CW, Zhang R (1987) Cytokinin translocation and metabolism in lupin species I. Zeatin riboside introduced into the xylem at the base of Lupinus angustifolius stems. Aust J Plant Physiol 14:695–718
Jones RJ, Schreiber BMN (1997) Role and function of cytokinin oxidase in plants. Plant Growth Regul 23:123–134
Kakimoto T (2001) Identification of plant cytokinin biosynthetic enzymes as dimethylallyl diphosphate: ATP/ADP isopentenyltransferases. Plant Cell Physiol 42:677–685
Kelly MG (2006) Characterizing genotype-specific differences in survival, growth, and reproduction for field grown, rapid cycling Brassica rapa. Environ Exp Bot 55:61–69
Kopečný D, Tarkowski P, Majira A, Bouchez-Mahiout I, Nogué F, Laurière M, Sandberg G, Laloue M, Houba-Hérin N (2006) Probing cytokinin homeostasis in Arabidopsis thaliana by constitutively overexpressing two forms of the maize cytokinin oxidase/dehydrogenase 1 gene. Plant Sci 171:114–122
Kudo T, Kiba T, Sakakibara H (2010) Metabolism and long-distance translocation of cytokinins. J Integr Plant Biol 52:53–60
Lara MEB, Garcia MG, Fatima T, Ehne R, Lee TK, Proels R, Tanner W, Roitscha T (2004) Extracellular invertase is an essential component of cytokinin-mediated delay of senescence. Plant Cell 16:1276–1287
Letham DS (1963) Regulators of cell division in plant tissue. New Zeal J Bot 24:336–350
Ma Q, Wang XM, Wang ZM (2008) Expression of isopentenyl transferase gene controlled by seed-specific lectin promoter in transgenic tobacco influences seed development. J Plant Growth Regul 27:68–76
Massonneau A, Houba-Hérin N, Pethe C, Madzak C, Falque M, Mercy M, Kopečný D, Majira A, Rogowsky P, Laloue M (2004) Maize cytokinin oxidase genes: differential expression and cloning of two new cDNAs. J Exp Bot 55:2549–2557
Miyawaki K, Kakimoto T, Matsumoto-Kitano M (2004) Expression of cytokinin biosynthetic isopentenyltransferase genes in Arabidopsis: tissue specificity and regulation by auxin, cytokinin and nitrate. Plant J 37:128–138
Miyawaki K, Tarkowski P, Matsumoto-Kitano M, Kato T, Sato S, Tarkowska D, Tabata S, Sandberg G, Kakimoto T (2006) Roles of Arabidopsis ATP/ADP isopentenyl transferases and tRNA isopentenyl transferases in cytokinin biosynthesis. Proc Natl Acad Sci USA 103:16598–16603
Morris RO, Bilyeu KD, Laskey JG, Cheikh NN (1999) Isolation of a gene encoding a glycosylated cytokinin oxidase from maize. Biochem Biophys Res Commun 255:328–333
Motyka V, Faiss M, Strnad M, Kamínek M, Schmülling T (1996) Changes in cytokinin content and cytokinin oxidase activity in response to derepression of ipt gene transcription in transgenic tobacco calli and plants. Plant Physiol 112:1035–1043
Musgrave M (2000) Realizing the potential of rapid-cycling Brassica as a model system for use in plant biology research. J Plant Growth Regul 19:314–325
Noodén LD, Leopold AC (1978) Phytohormones and the endogenous regulation of senescence and abscission. In: Letham DS, Goodwin PB, Higgins TJV (eds) Phytohormones and related compounds: a comprehensive treatise, vol II. Elsevier/North-Holland Biomedical Press, Amsterdam, pp 329–369
Noodén LD, Letham DS (1984) Translocation of zeatin riboside and zeatin in soybean explants. J Plant Growth Regul 2:265–279
Porra R (2002) The chequered history of the development and use of simultaneous equations for the accurate determination of chlorophylls a and b. Photosynth Res 73:149–156
Sakakibara H (2006) Cytokinins: activity, biosynthesis, and translocation. Annu Rev Plant Biol 57:431–449
Schmülling T, Bartrina Y, Manns I, Krupková E, Werner T (2003) Structure and function of cytokinin oxidase/dehydrogenase genes of maize, rice, Arabidopsis and other species. J Plant Res 116:241–252
Singh S, Bandendoch-Jones J, Jameson PE, Letham DS, Parker C, Zhang R, Nooden LD (1988) Cytokinin biochemistry in relation to leaf senescence. Plant Physiol 88:788–794
Song J, Clemens J, Jameson PE (2008) Quantitative expression analysis of the ABC genes in Sophora tetraptera, a woody legume with an unusual sequence of floral organ development. J Exp Bot 59:247–259
Takei K, Sakakibara H, Sugiyama T (2001) Identification of genes encoding adenylate isopentenyltransferase, a cytokinin biosynthesis enzyme, in Arabidopsis thaliana. J Biol Chem 276:26405–26410
Takei K, Ueda N, Aoki K, Kuromori T, Hirayama T, Shinzaki K, Yamaya T, Sakakibara H (2004) AtIPT3 is a key determinant of nitrate-dependent cytokinin biosynthesis in Arabidopsis. Plant Cell Physiol 45:1053–1062
Vyroubalova S, Vaclavikova K, Tureckova V, Novak O, Smehilova M, Hluska T, Ohnoutkova L, Frébort I, Galuszka P (2009) Characterization of new maize genes putatively involved in cytokinin metabolism and their expression during osmotic stress in relation with cytokinin levels. Plant Physiol 151:433–447
Werner T, Motyka V, Laucou V, Smets R, Van Onckelen H, Schmülling T (2003) Cytokinin-deficient transgenic Arabidopsis plants show multiple developmental alterations indicating opposite functions of cytokinins in the regulation of shoot and root meristem activity. Plant Cell 15:2532–2550
Williams PH, Hill GB (1986) Rapid-cycling populations of Brassica. Science 232:1385–1389
Zalewski W, Galuszka P, Gasparis S, Orczyk W, Nadolska-Orczyk A (2010) Silencing of the HvCKX1 gene decreases the cytokinin oxidase/dehydrogenase level in barley and leads to higher plant productivity. J Exp Bot 61:1839–1851
Acknowledgments
We acknowledge support from the Foundation for Research Science and Technology through LINX0803, Advanced Seed Production Systems.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
O’Keefe, D., Song, J. & Jameson, P.E. Isopentenyl Transferase and Cytokinin Oxidase/Dehydrogenase Gene Family Members are Differentially Expressed During Pod and Seed Development in Rapid-cycling Brassica . J Plant Growth Regul 30, 92–99 (2011). https://doi.org/10.1007/s00344-010-9171-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00344-010-9171-y