
Abstract
A new cellulose solution was prepared by a new cellulose solvent, tetrabutylammonium acetate (TBAA)/dimethyl sulfoxide (DMSO). A new kind of regenerated cellulose fibers was spun successfully for the first time from the cellulose solution by a wet spinning system. The dissolving process of cellulose in TBAA/DMSO was observed by confocal laser scanning microscope. The rheological of the cellulose dopes was determined by rotated rheometer, and the results showed that the cellulose/TBAA/DMSO solution was a typical shear-thinning fluid. In addition, the morphology, chemical structure and mechanical properties of the prepared cellulose fibers were characterized by scanning electron microscope (SEM), 13C CP/MAS NMR, Fourier transform infrared spectroscopy (FT-IR), X-ray diffraction (XRD) and electronic tensile tester, respectively. The SEM patterns showed that the regenerated fibers possessed a smooth surface and circular cross section. The results from 13C NMR and XRD patterns indicated that the novel fibers mainly exhibited amorphous cellulose. Meanwhile, the novel fibers have good mechanical properties.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
In this century, along with the oil resource exhausting and environmental pollution caused by petroleum chemical industry, scientific research works have moved toward renewable raw materials and more environmentally friendly resources [1, 2]. Biopolymers from various natural resources such as cellulose, starch and proteins have been taken for alternative materials to solve the widespread dependence on fossil fuel [2, 3] owing to they are abundant, renewable, biodegradable, and eco-friendly. According to the Technology Road Map for Plant/Crop-Based Renewable Resources 2020, sponsored by the US Department of Energy (DOE), it has the target to achieve 10 % of the basic chemical building blocks by 2020, and to 50 % by 2050 [3, 4]. From the present point of view, cellulose is the most common abundant renewable organic polymer and has a wide range of applications in fibers, films and plastics [5–7]. Moreover, numerous new functional materials about cellulose are being developed. As generally known, wood pulp remains the most important raw material source for the processing of cellulose [8]. However, most of which is used for the production of paper and cardboard. Only very small amounts of wood pulp were used for the production of cellulose regenerated fibers and films, as well as for the synthesis of cellulose esters and ethers. So, cellulose resources have a very good application prospect as a chemical raw material in the future [9].
Researching dissolution of cellulose and making use of cellulose to spin fibers has a long history in human history [10]. Regeneration of cellulose fibers is one of the most important tasks for the application of the native cellulose. Up to now, various methods are in use to regenerate cellulose fibers, such as the viscose [11], cuprammonium [12] and Lyocell processes [13]. The earliest technology for regeneration of cellulose fibers is the traditional viscose process. The viscose process unfortunately generates several environmentally hazardous byproducts, including CS2, H2S [10, 14], hence, requires additional facilities to deal with the gaseous and aqueous waste emissions. The other traditional way for preparing cellulose rayon is cuprammonium method. The cuprammonium method also has environmental problems and is only used to detect the degree of polymerization of polymer nowadays [11, 12]. Throughout the years, to minimize the hazardous byproducts as well as to simplify the processing steps, numerous new solvents for cellulose have been researched. A new solvent, namely a NaOH/thiourea aqueous solution, has been developed. The advantage of the solvent system is that has no pollution to environment. However, in this process, the dissolution of cellulose needs to undergo a freezing-to-thawing process, which prevents its industrialization. The dissolving capacity of the NaOH/thiourea aqueous solution is limited, which can only dissolve the cellulose of low degree of polymerization [10]. The N-methylmorpholine-N-oxide (NMMO) as an environmentally friendly process of cellulose-fiber spinning to replace the viscose process has become a more desirable method, leading to a new class of man-made cellulose fibers under the generic name of Lyocell (Biganska et al. 2008). Lyocell fibers show better performance qualities, whereas the Lyocell process still has many environmental issues that need to be overcome. These issues include the minimization of the undesired byproducts, efficient recovery of the expensive solvent [15], uncontrolled thermal stability of the system NMMO-cellulose-H2O and high tendency of fibrillation of the Lyocell fiber [1]. Phosphoric acid (PA) is one of the favored solvents for cellulose dissolution. The acid represents common mineral acids, such as orthophosphoric acid (OPA), pyrophosphoric acid (PPA), tetraphosphoric acid (TPA), and so on. Phosphoric acids as a potent solvent for cellulose, with or without additives such as organic acids, were patented already back in 1927. The composition of the acid is usually expressed in P2O5 concentration, which, at a concentration exceeding 74 % is anhydrous. Thus, mixing different species of phosphoric acids may give a powerful cellulose solvent, claimed to rapidly dissolve up to 38 % w/w cellulose [16]. In recent years, a new way of processing cellulose by using a new kind of green cellulose solvent: an ionic liquids (ILs) [17]. In 2002, imidazolium-based ionic liquids were found to dissolve cellulose. As ionic liquids are direct solvents for cellulose and have many attractive properties such as chemical and thermal stability, excellent dissolving capability, immeasurably low vapor pressure and recyclability, so that ionic liquids are considered desirable green solvents [18–21]. However, ionic liquids have internal drawbacks such as high viscosity and high water absorption. The reaction conditions of processing cellulose in ionic liquids are little harsh with higher reaction temperature, longer reaction time and strong agitation. Due to various reasons, ionic liquids have not been applied to industrial production. The necessity to face present problems, such as a shortage of fossil fuels, environmental changes and a rapidly growing population, has attracted extensive research to find a suitable solvent for dissolving cellulose from natural biomaterials, such as wood. If a new kind of regenerated cellulose fibers, films and other products could be obtained by using the novel cellulose solvent system, it would have a paramount and important impact on the cellulose chemical industry.
In this article, a new solvent was found in our laboratory to dissolve cellulose, including tetrabutylammonium acetate (TBAA) and dimethyl sulfoxide (DMSO). The new cellulose dissolving system and dissolution technique have the advantages of high efficiency, low-energy and pollution-free, compared with the existing cellulose solvents. However, the dissolution mechanism of TBAA/DMSO is studying and not fully elucidated. We deduce that the dissolution mechanism of this new solvent is similar to the TBAF·3H2O/DMSO. DMSO cannot dissolve cellulose alone while are the good swelling agents for polymer due to their bipolarity. According to previous research, the fluoride salt is the only halide salt of TBA+ capable of rendering cellulose soluble in DMSO [22]. Thus, we deduce that the acetate ions play a critical role in dissolving cellulose. With the strongly electronegative of acetate ions, it is a very strong hydrogen bond acceptor, favorably competes with the hydroxyl and acetal oxygen atoms of cellulose,thus breaking the network of hydrogen bonds between the cellulose chains. And yet, further researches are required to completely elucidate the dissolution mechanism of cellulose in the TBAA/DMSO. Additional, much work has been done on the recycling solvent. The method used at present is reduced pressure distillation and extraction. We believe that the novel solvent will create an interesting and important challenge in cellulose industry. In our work, we attempted to prepare a new kind of regenerated cellulose fibers from the TBAA/DMSO solution by wet spinning with an ethanol bath. TBAA and DMSO have larger solubility in the ethanol, which is available for regenerating of cellulose.
2 Experimental
2.1 Materials
The cellulose (soft wood pulp) with α-cellulose content of more than 92 % was provided by the Shandong Rizhao Senbo Pulp Paper Co., Ltd., China. The viscosity-average molecular weight (M w) of the cellulose sample was determined to be 17.01 × 104 g mol−1 (DP = 1,050) determined by viscometry. The soft wood pulp was vacuum-dried for 24 h at 60 °C to remove any moisture before use. The new solvent TBAA was purchased from TCI, Shanghai. DMSO, NaOH, ethanol, ethylenediamine and other chemicals were of analytical reagent and used without further purification.
2.2 Preparation of cellulose solutions
The new cellulose solutions were prepared with TBAA/DMSO. A certain amount of TBAA/DMSO was put into 50 ml bottle, and the bottle was heated to 40 °C. Then, an appropriate amount of dried cellulose was added to the bottle, and the mixture was stirred continually for 1 h. The cellulose solutions of 4, 6, 8 and 10 wt% in the new solvent were obtained. A high-vacuum pump was used to remove air bubbles in the solutions at 40 °C for 2 h.
The resulting cellulose solutions were analytically characterized and used for a further spinning process.
2.3 Spinning experiment
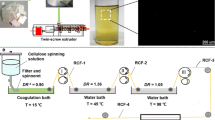
A homemade wet spinning equipment, which is consisting of a micro-syringe pump system, a glass syringe (inner diameter, 10 mmΦ) with a needle (length, 35 mm; inner diameter, 0.355 mmΦ) as a spinneret, a spinning bath, a turning round godet, a take-up device, was used for the shaping trials. The cellulose solution obtained earlier was introduced into glass syringe with a precise drive of the piston. The extrusion speed is 0.5 ml/min. The dopes temperature can be exchange by the heating cylinder. Temperature of the spinning dope and coagulation bath is 50 and 25 °C, respectively.
3 Methods
3.1 Pulp characterization
3.1.1 Degree of polymerization (DP) characterization
The viscosity-average degree of polymerization (DP) of the used pulp and regenerated fibers was characterized concerning intrinsic viscosities in [η]Cuen according DIN 54270. Part 2. DP was determined in cupriethylenediamine by means of ubbelohde viscometer at 25 °C. The DP was calculated according to the Eq. (1):
The content of α-cellulose in the used pulp was determined as the remaining pulp that was no dissolved in a 17.5 wt% NaOH at 20 °C.
3.2 The dissolution of cellulose in TBAA/DMSO
The dissolution process of cellulose in TBAA/DMSO was observed by confocal laser scanning microscope (CLSM) (Leica TCS SP5, Germany) with a hot stage and a multicolor digital camera. A small quantity of soft wood pulp and TBAA/DMSO solvent was sandwiched between two pieces of cover glasses and put on the hot stage at 40 °C.
3.3 The spinning dopes characterization
Rheological properties of the spinning dopes were determined using a Bohlin instruments CVO-100-901 rheometer equipped with a pp20 plate measuring system for both viscometry and oscillatory measurements.
3.4 Fiber characterization
3.4.1 SEM
The surface and cross section of fibers spun from the cellulose concentration of 8 wt% were observed by means of scanning electron microscope (S-3400 N, HITACHI). The fibers were frozen in liquid nitrogen, immediately snapped and then vacuum-dried. The samples was prepared and sputtered with gold before observed and photographed.
3.4.2 NMR
13C NMR spectra were recorded on Ultrashield Plus 400 spectrometer (Bruker Avance III, Germany, magnetic field = 9.4 T, 13C frequency = 100.38 MHz) spectrometer with a (CP/MAS) unit at room temperature. The spinning rate and the contact time were 5.0 kHz and 3.0 ms, respectively.
3.4.3 FT-IR
FTIR spectra of cellulose and regenerated cellulose fibers were the result of averaging 32 scans in wavelength ranging from 4,000 to 500 cm−1 at a resolution of 2 cm−1 by using dry powdered samples on an infrared spectrometer (Tensor 27, Bruker, Germany). Pellets were prepared by combining the samples with KBr.
3.4.4 XRD
X-ray diffraction measurements of the regenerated fibers were performed on a XRD diffractometer (Shimadzu XRD-6000, Japan). Samples were vacuum-dried for 24 h before measurement and cut into powders so as to erase the influence from the crystalline orientation of each fiber sample [23]. The patterns measured with Cu Kα radiation (λ = 0.154 nm) at 40 kV and 30 mA. The powder samples were scanned from 5° to 40° at a scanning rate of 2°/min.
3.4.5 The mechanical properties of the regenerated fibers
Denier of fibers was estimated assuming that the cross section of the fiber is a true circle, by weighting the dried fiber with 80 cm in length. The tensile strength and elongation mechanical of the dried fibers were measured on an electronic tensile tester (Instron 5848, Germany). The sample length was 20 mm. Each specimen was tested for 20 times and took the average.
4 Results and discussion
4.1 Morphology of cellulose in the TBAA/DMSO at 40 °C
The dissolving process of cellulose in the new solvent was observed by CLSM. As shown in Fig. 1, the cellulose with a DP values as high as 1,050 could dissolve in the TBAA/DMSO within 30 min at 40 °C. Such rapid dissolution rate in a solvent for cellulose was only reported in the TBAF/DMSO solvent system [22]. For the 1-butyl-3-methylimidazolium (BMIMCl) solvent, with the temperature further going up to 80 °C, the cellulose began to dissociate and dissolved gradually [21]. Assuming the technology can apply to industrialization production, it is significant that the cellulose dissolved in TBAA/DMSO rapidly without any pretreatment or activation at 40 °C.

CLSM images of cellulose dissolution in TBAA/DMSO at different times (DP = 1,050)
4.2 Steady shear rheological behaviors of cellulose/TBAA/DMSO solutions
As shown in Fig. 2, the cellulose/TBAA/DMSO solutions exhibited a character of typical shear-thinning behavior with variation of shear rates. This behavior is similar to that of other cellulose solutions such as in NMMO [24] and [BMIM]Cl [5]. The shear viscosity values became larger with the increase in concentration. It indicated that the chain entanglements of cellulose increased as the cellulose concentration increases.

Steady shear rheological curves for the cellulose/TBAA/DMSO solution at 50 °C
4.3 Dynamic oscillation behaviors of cellulose/TBAA/DMSO solutions
The relationship between storage modulus G′ and loss modulus G″ will provide more information about the polymer interaction. As long as loss modulus G″ larger than the storage modulus G′, the system is deemed to a fluid, while the opposite is true for a semisolid such as a gel [25]. Figure 3 shows the storage modulus G′ and loss modulus G″ curves as a function of angular frequency for cellulose solutions. As the cellulose concentration increases from 4 to 10 %, the G′ and G″ values tend to become larger. It shows that the ability of temporary networks was enhanced by an increase in cellulose solution. This can be contributed to that the segment density rises and the number of entanglement points increases with the increase in cellulose concentration.

Modulus of cellulose/TBAA/DMSO solutions with different concentration at 50 °C
In this situation, the G′ < G″ behavior only occurs at low frequencies. This can be explained that the cellulose chains have time to avoid the externally imposed deformation. The deformation takes place so slowly in cellulose solution that the majority of the energy is dissipated by viscous flow. With the increase in frequency, G′ increases more sharply than does the G″, which leads to a crossover point. As the concentration increases, the crossover point G′(w) = G″(w), shifts to lower values. It shows that the ability of the entanglement networks was enhanced with the increase in polymer concentrations. It can be attributed to that the molecular chains of cellulose are more close to each other at higher concentration.
4.4 Morphology of the fibers
Figure 4 shows the surface and cross section of the novel cellulose fibers observed by scanning electron microscopy. The novel cellulose fibers had a smooth surface without obvious crevices or flaws. In addition, the novel fibers had a circular cross section, which was similar to that of Lyocell fibers and ionicell fibers [4]. A crack was can be seen in the cross section of novel fibers, this turn of phenomenon is caused by the brittle fracture.

scanning electron microscopy photographs of surfaces (a) and cross sections (b) of the novel cellulose fibers
4.5 13C CP/MAS NMR spectra
Figure 5 shows the 13C NMR spectrum of cellulose dissolved in TBAA/DMSO. The spectrum of cellulose solution exhibited six singlets at 103.5, 79.4, 76.4, 75.6, 74.2 and 60.8 ppm, assigned to the C1, C4, C5, C3, C2 and C6. No additional resonances appear in the 13C NMR spectrum of the cellulose solution, indicating there is no derivate reaction during the process of cellulose dissolution in the novel solvent. The spectrum is very similar to that of cellulose dissolved in other solvents like TBAF [22] and BMIMCl [26]. Therefore, TBAA/DMSO can be considered as a non-derivate solvent for cellulose.

13C NMR spectrum of cellulose dissolved in TBAA/dimethyl sulfoxid-d 6
Figure 6 shows the 13C CP/MAS spectra of soft wood pulp and novel fibers. The spectrum of soft wood pulp consisted of the following resonances: 104.4 (C-1), 88.7 (C-4), 74.6 (C-5, C-3), 72.0 (C-2) and at 65.0 ppm (C-6), which were assigned to cellulose. The signals at 88.7 and 65.0 ppm correspond to the C-4 and C-6 of crystalline cellulose [27]. The broad shielding shoulders appear in the C4 regions of the novel fibers, corresponding to peaks in the amorphous region [28]. This indicated that the crystallinity value dramatically decreased due to the dissolution and regeneration treatment processes. The C6 peak of novel fibers was located at 62.3 ppm shifted to higher magnetic fields than the native cellulose (65.0 ppm), revealing that “t-g” conformation of C6–OH group (for cellulose I) had changed into a “g-t” conformation (for cellulose II) with the simultaneous formation of intramolecular hydrogen bonds O6–H–O ′2 [29]. Due to the flaws of the homemade spinning machine, the regenerated cellulose fibers could not be oriented well along the axis. Therefore, the novel fibers should contain larger amount of amorphous region and a small amount of crystalline region.

Cross-polarization/magic-angle spinning (CP/MAS) 13C NMR spectrum of soft wood pulp and novel fibers
4.6 FT-IR spectral
FT-IR spectral of original cellulose and regenerated cellulose fibers were shown in Fig. 7. It showed that the two spectral were quiet similar, and no new peaks appeared in the regenerated cellulose fibers. This phenomenon indicated that no chemical reaction occurred during the dissolution and coagulation processes of cellulose. A broad vibration band was located in the range of 3,000–3,500 cm−1, which represented the O–H vibrations of cellulose. Compared with original cellulose, the bank of the O–H vibration of the regenerated cellulose fibers shifted to a higher frequency and became sharper and narrower. This could be explained that there generated more O–H vibration in regenerated cellulose fibers for splitting hydrogen bonds [30]. The strong absorption peak at 1,050 cm−1 belonged to the frame vibration of the glycosidic bond of cellulose (C–O–C). Compared with original cellulose, the bank of the C–O–C vibration of the regenerated cellulose fibers became broader and gentler. This indicated that the C–O–C bonds of cellulose were broken down in the process of dissolving.

FT-IR spectral of original cellulose and regenerated cellulose fibers: a original cellulose b regenerated cellulose fibers
.
4.7 Crystal structure of the fibers
Figure 8 shows the X-ray diffraction patterns of the soft wood pulp and novel cellulose fiber. It was shown that the XRD profiles of soft wood pulp exhibited typical cellulose I angles (2θ) around at 14.9°, 16.8° and 22.8°, which are assigned to the Miller indices of (1–10) (110) and (020) [31]. In comparison, the peak from regenerated cellulose fibers at approximately 2θ = 20.2° is attributed to the two diffraction planes of (110) and (020) due to the overlap into one peak [32]. The result showed that the crystal structure of the regenerated cellulose was destroyed and most of crystalline cellulose converted into amorphous cellulose. Therefore, the XRD result was consistent with the NMR.

X-ray diffraction patterns of the soft wood pulp and novel cellulose fiber
4.8 Properties of the novel cellulose fiber
The \(\overline{DP}\), denier, tensile strength, elongation at break of the novel fibers were summarized in Table 1. The \(\overline{DP}\) value of the novel fibers determined were 975, close to that of the starting soft wood pulp material. The result indicated that no obvious degradation of cellulose did occur in the dissolution and regeneration process. With concentration of the novel cellulose solution increase from 4 to 10 wt%, the tensile strength varies from 2.15 to 3.07 cN/dtex. This can be explained that the segment density rises and the number of entanglement points increases with the increase in cellulose concentration. Laszkiewicz et al. [33] have reported that the tensile strength of viscose fibers depends not only on the cellulose content, but also on the draw ratio of fibers during spinning. Therefore, the mechanical properties of the novel fibers can be enhanced with different draw ratios.
5 Conclusions
In our study, a new cellulose solvent, TBAA/DMSO, was used for dissolving cellulose pulp for the first time. The novel regenerated cellulose fibers were prepared successfully from the spinning dope by wet spinning equipment. Without the formation of cellulose derivatives that indicated the TBAA/DMSO solvent was a direct dissolution for cellulose. The rheological behavior of the cellulose/TBAA/DMSO solutions showed that the cellulose/TBAA/DMSO solution was a typical shear-thinning fluid. Moreover, the novel fibers had a smooth surface as round and compact structure. Therefore, the TBAA/DMSO can provide a new way for dissolving cellulose and the novel cellulose fibers could be a new kind of environment friendly cellulose fiber.
References
J. Cai, L. Zhang, J. Zhou, H. Qi, H. Chen, T. Kondo, X. Chen, B. Chu, Adv. Mater. 19, 821 (2007)
Y. Yao, K.S. Mukuze, Y. Zhang, H. Wang, Cellulose 21, 675 (2013)
D. Klemm, B. Heublein, H.P. Fink, A. Bohn, Angew. Chem. Int. Ed. 44, 3358 (2005)
T. Cai, H. Zhang, Q. Guo, H. Shao, X. Hu, J. Appl. Polym. Sci. 115, 1047 (2010)
X. Chen, Y. Zhang, L. Cheng, H. Wang, J. Polym. Environ. 17, 273 (2009)
D. Ruan, L. Zhang, A. Lue, J. Zhou, H. Chen, X. Chen, B. Chu, T. Kondo, Macromol. Rapid Commun. 27, 1495 (2006)
S. Iwamoto, A. Isogai, T. Iwata, Biomacromolecules 12, 831 (2011)
P. Chen, H. Yu, Y. Liu, W. Chen, X. Wang, M. Ouyang, Cellulose 20, 149 (2012)
Q. Li, J. Zhou, L. Zhang, Cellulose 19, 1547 (2012)
D. Ruan, L. Zhang, J. Zhou, H. Jin, H. Chen, Macromol. Biosci. 4, 1105 (2004)
H.P. Fink, P. Weigel, H.J. Purz, J. Ganster, Prog. Polym. Sci. 26, 1473 (2001)
X. Chen, C. Burger, D. Fang, D. Ruan, L. Zhang, B.S. Hsiao, B. Chu, Polymer 47, 2839 (2006)
O. Biganska, P. Navard, Cellulose 16, 179 (2008)
S. Ghittori, L. Maetri, I. Contardi, P. Zadra, P. Marraccini, M. Imbriani, Am. J. Ind. Med. 33, 478 (1998)
T. Rosenau, A. Potthast, P. Kosma, Adv. Polym. Sci. 205, 153 (2006)
M.G. Northolt, H. Boerstoel, H. Maatman, R. Huisman, J. Veurink, H. Elzerman, Polymer 42, 8249 (2001)
R.P. Swatloski, S.K. Spear, J.D. Holbrey, R.D. Rogers, J. Am. Chem. Soc. 124, 4974 (2002)
S. Zhu, Y. Wu, Q. Chen, Z. Yu, C. Wang, S. Jin, Y. Ding, G. Wu, Green Chem. 8, 325 (2006)
B. Kosan, C. Michels, F. Meister, Cellulose 15, 59 (2007)
A. Pinkert, K.N. Marsh, S. Pang, M.P. Staiger, Chem. Rev. 109, 6712 (2009)
G. Jiang, W. Huang, B. Wang, Y. Zhang, H. Wang, Cellulose 19, 679 (2012)
T. Heinze, R. Dicke, A. Koschella, A.H. Kull, E.-A. Klohr, W. Koch, Macromol. Chem. Phys. 201, 627 (2000)
C. Yamane, M. Mori, M. Saito, K. Okajima, Polym. J. 28, 1039 (1996)
D.W. Chae, B.C. Kim, W.S. Lee, J. Appl. Polym. Sci. 86, 216 (2002)
C. Olsson, G. Westman, J. Appl. Polym. Sci. 127, 4542 (2013)
H. Zhao, J. Kwak, Y. Wang, J.A. Franz, J.M. White, J.E. Holladay, Carbohydr. Polym. 67, 97 (2007)
X. Liu, J.H. Pang, X.M. Zhang, Y.Y. Wu, R.C. Sun, Cellulose 20, 1391 (2013)
J. Cai, L. Zhang, J. Zhou, H. Li, H. Chen, H. Jin, Macromol. Rapid Commun. 25, 1558 (2004)
J.A. Cuculo, C.B. Smith, U. Sangwatanaroj, E.O. Stejskal, S.S. Sankar, J. Polym. Sci. A Polym. Chem. 32, 241 (1994)
H. Zhang, J. Wu, J. Zhang, J. He, Macromol 38, 8272 (2005)
A.D. French, Cellulose 21, 885 (2013)
A.D. French, M.S. Cintron, Cellulose 20, 583 (2013)
B. Laszkiewicz, P. Wcislo, J.A. Cuculo, J. Appl. Polym. Sci. 46, 445 (1992)
Acknowledgments
This work was supported by the project of the state forestry administration (948, 2013-4-03).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Sun, H., Miao, J., Yu, Y. et al. Dissolution of cellulose with a novel solvent and formation of regenerated cellulose fiber. Appl. Phys. A 119, 539–546 (2015). https://doi.org/10.1007/s00339-015-8986-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00339-015-8986-6