
Abstract
Body colour has played a significant role in the evolution of coral reef fishes, but the phylogenetic level at which colour variation is expressed and the evolutionary processes driving the development and persistence of different colour patterns are often poorly understood. The aim of this study was to examine the genetic relationships between multiple colour morphs of Pseudochromis fuscus (family Pseudochromidae), both within and among geographic locations. Pseudochromis fuscus is currently described as a single species, but exhibits at least six discrete colour morphs throughout its range. In this study, P. fuscus from Papua New Guinea (PNG) and the Great Barrier Reef (GBR), Australia, formed three genetically distinct clades based on mitochondrial DNA (control region) sequence data: (1) yellow and brown morphs from the GBR and southern PNG, as well as an orange morph from southern PNG; (2) a pink morph from southern PNG; and (3) all three morphs (pink, orange and grey) found in Kimbe Bay, northern PNG. The three groups showed deep levels of divergence (d=14.6–25.4%), suggesting that P. fuscus is a complex of polychromatic species, rather than a single widespread species with many different colour morphs. Population genetic analyses indicate that the three clades have experienced unique evolutionary histories, possibly from differential effects of sea level fluctuations, barriers to gene flow and historical biogeography.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Coral reef fishes exhibit extreme diversity in colouration, yet the phylogenetic level at which differences in colouration are expressed and the evolutionary mechanisms that produce this extraordinary diversity in colour are poorly understood (McMillan et al. 1999; Bernardi et al. 2002). Colour patterns play an important role in the identification of coral reef fishes (e.g. Lieske and Meyers 1999) and many species are recognised as having two or more different colour morphs (e.g. Fischer 1980; DeMartini and Donaldson 1996; Planes and Doherty 1997; Taylor and Hellberg 2002; Munday et al. 2003). In some cases different colour morphs occur sympatrically, in other instances the morphs occur in different geographic locations. The presence of different colour morphs in otherwise morphologically similar animals provides a challenge for traditional taxonomy. It is possible that differently coloured morphs, occurring either in sympatry or allopatry, are reproductively isolated and should be treated as separate species. Alternatively, different colour morphs might simply be variants of a single polychromatic species.
Some colour variants exhibit clear genetic differentiation. For example, there is deep genetic divergence between three colour forms of the cleaner goby, Elacatinus evelynae, found on neighbouring Caribbean Islands (Taylor and Hellberg 2002). Colour variants of other species exhibit genetic differentiation both between morphs and between geographic locations. For example, different colour morphs of the spiny damselfish, Acanthochromis polyacanthus, found on the same reef can be distinguished genetically (Planes and Doherty 1997) and these same colour morphs also exhibit genetic differences between widespread locations (van Herwerden and Doherty 2005). In contrast, hamlets of the genus Hypoplectrus exhibit a number of genetically indistinguishable colour morphs at various locations in the Caribbean (Ramon et al. 2003; Garcia-Machado et al. 2004), which have been described as separate species by some authors, while others regard them as colour variants of a single species (Thresher 1978; Fischer 1980; Domier 1994). These examples demonstrate the potential genetic complexity that can occur between colour morphs, both within and between populations.
Genetic divergence can also be found between geographically separated populations, independently of differences in colouration. For example, geographic location rather than colour pattern was the best predictor of genetic isolation in the Dascyllus trimaculatus species complex (Bernadi et al. 2002) and significant population differentiation has been found in a range of marine fishes that do not possess colour polymorphisms (reviewed by Planes 2002; also Rocha et al. 2002; Florence et al. 2002; Planes and Fauvelot 2002; Thacker 2004). Genetic structure between locations can be influenced by life-histories and behaviour of the dispersive phase, ocean currents, and the distance between populations (Palumbi 1994; Ward et al. 1994; Doherty et al. 1995; Bohonak 1999). However, the often inconsistent relationship between genetic structure and the potential for dispersal indicates that, in many instances, historical biogeography and vicariance events also play an important role in explaining present-day population structures (Avise 1998; Bellwood and Wainwright 2002; Planes and Fauvelot 2002). Overall, a combination of factors are often likely to be involved in generating genetic differentiation between populations (Avise 2000; Muss et al. 2001; Planes 2002).
Pseudochromis fuscus (family Pseudochromidae) is a polychromatic species with a wide distribution throughout the Indo-Pacific. Its range stretches as far as Sri Lanka in the west, southern Japan in the north, Vanuatu in the east and the southern Great Barrier Reef in the south (Gill 2004; Michael 2004). Pseudochromis fuscus exhibits a range of colour variants, but little other morphological variation, and it is unclear whether this is a single species with many colour morphs or a complex of separate species. At least six different colour morphs of P. fuscus have been described (Gill 2004; Michael 2004), but more may exist. Some colour morphs appear to be geographically restricted, whereas others are widely distributed. Furthermore, some populations appear to be represented by a single colour morph, whereas other populations contain multiple colour variants. The taxonomic status of P. fuscus is further complicated by the existence of sex-limited colour morphs (Eyre 2001) as well as apparently ecologically based forms that are not sex-limited (Munday et al. 2003). An ontogenetic basis has not been suggested for any of the colour variants. The taxonomic uncertainty of P. fuscus has not been resolved using traditional approaches and all colour morphs are currently pooled into a single species.
The aim of this study was to apply, for the first time, molecular tools to examine the relatedness of different colour morphs and develop hypotheses concerning the mode of genetic diversification in the P. fuscus complex. Molecular tools have proven useful in other groups where traditional taxonomic approaches are difficult, or are likely to be uninformative (Palumbi 1997; Knowlton 2000; Colborn et al. 2001; Rocha 2004). A molecular phylogeographic analysis, using the mitochondrial control region (d-loop), was employed to determine whether different colour morphs of P. fuscus from the Great Barrier Reef (GBR) and Papua New Guinea (PNG) were genetically differentiated. The results of the genetic analysis were compared to morphometric data (fin ray counts) to test for concordance between morphological and molecular approaches. The genetic relationships between colour morphs of P. fuscus at four geographic locations separated by several thousand kilometres was also evaluated to help determine the relatedness of the different colour morphs and infer their evolutionary histories.
Materials and methods
Study species
The family Pseudochromidae contains approximately 80 valid species (Gill 2004), a number of which exhibit more than one colour variant. Colour morphs are sex-limited in some species (Eyre 2001; Gill 2004), but not others (Munday et al. 2003). Therefore, it appears that both sexual selection and ecology have been important to the evolution of colour variation in this group of fishes (Munday et al. 2003).
Pseudochromis fuscus is a small (maximum size ~10 cm total length) carnivorous species, common on shallow coral reefs in the western Pacific. Individuals are usually found near complex habitat, such as rubble or live coral (Munday et al. 2003; Michael 2004). On the Great Barrier Reef, the brown morph of P. fuscus is most commonly found on rubble habitat in association with brown damselfishes. In contrast, the yellow morph is most commonly found near live coral in association with yellow damselfishes (Munday et al. 2003). Individuals occupy small territories that they defend against conspecifics (personal observation). Spawning is demersal and males care for the eggs until they hatch (Michael 2004).
Genetic analysis
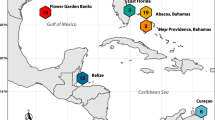
Samples of Pseudochromis fuscus were obtained from four geographic locations: Kimbe Bay in northern PNG (5°31′S, 150°5′E), Loloata Island in southern PNG (9°32′S, 147°17′E), Lizard Island on the northern GBR (14°40′S, 145°27′E) and One Tree Island on the southern GBR (23°30′S, 152°5′E) (Fig. 1). Two individuals of Pseudochromis marshallensis collected from Kimbe Bay were used as an outgroup for the phylogenetic analysis. Fish were collected on SCUBA between 2000 and 2003 using an anaesthetic (clove oil) and hand nets and were preserved in 80% or 100% alcohol. Five colour morphs were considered in this study. Their colour descriptions, distribution and biological characteristics, as well as the number of individuals used in the genetic analysis, are summarised in Table 1.

Map of Papua New Guinea and the Great Barrier Reef, Australia, showing the four sampling locations: One Tree Island, Lizard Island, Loloata Island and Kimbe Bay
PCR amplification
A standard salt-chloroform protocol was used to extract total DNA by proteinase K digestion from fin or muscle tissue (Sambrook et al. 1989). The 5′ end of the mitochondrial control region (d-loop) was PCR-amplified using universal primers L15995 (5′-AAC TCT CAC CCC TAG CTC CCA AAG-3′) and H16498 (5′-CCT GAA GTA GGA ACC AGA TG-3′) (Lee et al. 1995; Orbacz and Gaffney 2000). Amplification was performed in a 20 μl reaction volume containing 2.5 mM Tris pH 8.7, 5 mM KCl(NH4)SO4, 200 μM each dNTP, 3.5 mM MgCl2, 5 μM each primer, 0.5 units of Taq DNA Polymerase (Qiagen) and 10 ng template DNA. Touchdown PCR was performed with a total of 30 cycles divided into three phases consisting of 5, 5 and 20 cycles. Initial denaturation at 94°C was carried out for 2 min, followed by 5 cycles of denaturation for 30 s at 94°C, annealing for 30 s at 51°C and extension at 72°C for 90 s. A subsequent 5 cycles were performed as before, but with an annealing temperature of 49°C, followed finally by 20 cycles as before, but with 47°C annealing temperature. The profile was ended by a 10-min terminal extension phase at 72°C.
PCR products were purified using isopropanol precipitation and directly sequenced in forward and reverse directions using dye terminator 3.1 chemistry (ABI). The automated sequencing profile consisted of an initial denaturation at 96°C for 2 min, followed by 25 cycles of denaturation for 30 s at 96°C, annealing for 15 s at 50°C and extension for 4 min at 60°C. A 1°C per second ramp rate was used for all temperature changes during the cycle sequencing protocol. Sequenced products were cleaned by isopropanol precipitation and vacuum dried for 10 min. Sequences for 93 individuals of P. fuscus and 2 P. marshallensis were obtained from an ABI377 sequencer at Griffith University. All sequences were submitted to GenBank (Accession no. AY553417-AY553511).
Analysis of population genetic structure
Forward and reverse sequences were edited and aligned in Sequencher Version 4.1 (Gene Code Corporation). The sequences of all individuals were then aligned in Clustal X Version 1.8 (Thompson et al. 1997). Manual adjustments to the alignments were done by eye in Se-Al Version 1.0 alpha (Rambaut 2003).
Phylogenetic analysis
A first approximation of the phylogenetic relationships between morphs and geographic locations was inferred using distance and parsimony (heuristic search)-based methods in PAUP* 4.0b10 (Swofford 1998). To determine statistical support for branch nodes, 1,000 bootstrap pseudo-replications were performed.
A likelihood approach, implemented in Modeltest Ver. 3.06 (Posada and Crandall 1998) was used to determine which evolutionary model best fitted the data set. The HKY+G substitution model with a transition / transversion ratio of 2.9258, a proportion of invariable sites (I=0) and gamma distribution shape parameter (α=0.5609) was found to be the best fit model by both the hierarchical likelihood ratio tests (-Ln=2420.6104) and the Akaike Information Criterion (AIC=4851.2207). These parameters were then specified in a maximum likelihood analysis in PAUP* Ver. 4.0b10 (Swofford 1998) on a Unix super computer. The best likelihood tree was retained. Tree topologies of the maximum likelihood, distance and maximum parsimony analyses were found to be congruent, thus only the maximum likelihood tree will be presented. Majority rule consensus values were obtained from Bayesian inference of the data after discarding 21,000 out of 106 trees as burn-in. A haplotype network was constructed based on the minimum spanning tree (Rohlf 1973) implemented in Arlequin Ver. 2000 (Schneider et al. 2000). Nucleotide and haplotype diversities were estimated using equation 8.5 of Nei (1987) (analysis performed in Arlequin Ver. 2000).
Analysis of molecular variance
An Analysis of Molecular Variance (AMOVA) (Excoffier et al. 1992) was performed in Arlequin Ver. 2000 (Schneider et al. 2000) to test for hierarchical genetic variation within P. fuscus. A Markov chain analysis of 10,000 steps was used to estimate significance levels of the Φ statistics as per Raymond and Rousset (1995). The three fixation indices were defined as per Excoffier et al. (1992). Groups and populations were defined a priori based on colour, region and location.
Molecular clock
A relative rates test (Nei and Kumar 2000) was performed in MEGA2 Version 2.1 (Kumar et al. 2001) to test if a molecular clock could be applied to the d-loop data. The overall percentage of divergence was obtained by calculating the percentage of substitutions for each clade separately from the ancestral root node using the branch lengths from the maximum likelihood tree and HKY+G parameter genetic distance. The divergence rate used here was based on divergence rate estimates for the conserved sections (1.1%) as well as the hypervariable region (12.9%) of the mitochondrial control region (Alvarado et al. 1995). The overall divergence rate for the P. fuscus d-loop was calculated by adjusting for the proportion of conserved:hypervariable sites of the sequence data. Time since divergence was then calculated by dividing the percent divergence for each clade by the rate of divergence (percent per million years) and was compared to that of the mitochondrial control region for fish from the literature (Martin and Palumbi 1993; Stepien and Faber 1998; Donaldson and Wilson 1999; Bowen et al. 2001; Sturmbauer et al. 2001; Waters and Wallis 2001).
Morphology
The number of dorsal-fin rays in P. fuscus varies from 25 to 29 and has been identified as one of the most variable morphological characteristics exhibited by this species (Gill 2004). Segmented dorsal-rays were counted to determine if meristic characters supported the findings of the genetic analysis. The specimens used for fin-ray counts were selected from the collections for the genetic analysis based on their condition (intact dorsal fin).
Results
Genetic analysis
A 427 base pair fragment from the mitochondrial control region was sequenced for 93 individuals belonging to six morphs of P. fuscus from four different geographic locations. The sequences of the outgroup species P. marshallensis consisted of 543 base pairs. Excluding P. marshallensis from the analysis, 172 of the 427 sites were variable, of which 152 were parsimony informative. The transition (ts)/transversion (tv) ratio was 2.926. The nucleotide composition was AT-biased (A=35.19%, C=21.24%, G=14.47% and T=29.10%), which is commonly found in mitochondrial DNA of fishes (McMillan and Palumbi 1997).
Phylogenetic analysis
None of the 109 trees retained by the maximum likelihood analysis (-lnL ranged from 2411.487 to 2609.293) were significantly different (P>0.05) from the best tree (-lnL=2,411.487) (Shimodiara-Hasewaga test). The best tree did not differ from the bootstrap consensus tree with respect to assignment of individuals to the main clades and is presented here (Fig. 2). Three clades were identified, consisting of: (1) all three colour morphs (orange, pink and grey) from Kimbe Bay; (2) the pink morph from Loloata Island; and (3) the yellow and brown morphs from One Tree Island, Lizard Island and Loloata Island, as well as the orange morph from Loloata Island (referred to from here on as the GBR clade). High majority rule consensus and bootstrap values strongly supported all three clades of the tree.

Best maximum likelihood (ML) tree of 109 retained ML trees (100 bootstrap replicates) of mitochondrial control region sequences from 93 individuals of Pseudochromis fuscus. Two individuals of P. marshallensis were used as an outgroup. OT One Tree Island, LI Lizard Island, LOL Loloata Island and KB Kimbe Bay. Numbers above the lines indicate genetic distance. Numbers below the line are majority rule consensus values from the Bayesian inference and bootstrap support values from the maximum likelihood analysis, respectively
Six main results emerged from the phylogenetic analysis. First, no genetic differentiation between the yellow and brown morph from the GBR could be found using the mitochondrial control region as a molecular marker. Second, no genetic differentiation was found between colour morphs from Kimbe Bay (pink, orange and grey). Third, no genetic differentiation was evident between the locations on the GBR (One Tree Island and Lizard Island) and Loloata Island in southern PNG. Fourth, strong genetic divergence was found between Kimbe Bay in northern PNG versus Loloata Island in southern PNG and the two locations on the GBR. Fifth, the pink morph from Loloata forms a clade separate to the other morphs from the same location. Finally, the two PNG clades, Loloata Island pink morph and the Kimbe Bay morphs are more closely related (sisters), than either one is to the GBR clade (which also contains the remaining Loloata Island morphs).
Population genetic analysis
The haplotype network reinforced the phylogenetic tree by clustering the same individuals into three distinct groups (Fig. 3). The GBR morphs were separated from the Kimbe Bay morphs by 97 nucleotide substitutions and the Loloata Island pink morph was separated from the Kimbe Bay group by 88 nucleotide substitutions. Nucleotide substitutions between haplotypes within the three groups were relatively low to intermediate (1–3 substitutions separated haplotypes in the Loloata Island pink clade, 1–4 in the GBR clade and 2–12 in the Kimbe Bay clade). Seven of the thirty one haplotypes present in the GBR clade were shared by more than one individual, accounting for 35 of the 59 individuals sampled there. Five of fourteen haplotypes present in the Kimbe Bay clade were shared, accounting for 14 of the 23 individuals sampled there. In contrast, all individuals in the Loloata Island pink morph clade had unique haplotypes. A total of 57 haplotypes in the 93 individuals of P. fuscus were identified (Fig. 3). Haplotype diversities for each clade were high, whereas nucleotide diversities were variable (GBR: h=0.92, π=0.57%±0.35%; KB: h=0.92, π=2.98%±1.56%; LOL pink: h=1, π=0.82%±0.52%,). Overall haplotype and nucleotide diversities were very high (h=0.96, π=12.8%±6.2%).

Relationship among haplotypes from the mitochondrial control region of Pseudochromis fuscus presented in a minimum spanning haplotype tree. The different colour morphs from each location can be identified by different colour fills. The size of the circle indicates the number of individuals sharing the particular haplotype. Crossbars on the lines connecting haplotypes specify number of substitutions separating haplotypes. Overall nucleotide (π) and overall haplotype diversities (h) are shown
AMOVA
Based on the strong phylogenetic structure present in the data, three groups were defined a priori for the analysis of molecular variance (AMOVA): (1) GBR, (2) KB, and (3) LOL pink. Within groups 1 and 2, the populations were based on colour and location. The GBR group (1) contained OT yellow, OT brown, LI yellow, LI brown, LOL yellow, LOL brown, and LOL orange, whereas the Kimbe Bay group (2) included KB pink, KB orange, and KB grey. Both Φ ST and Φ CT values were found to be extremely high (0.9556 and 0.954, respectively) and highly significant (P=0.00000; 1,023 permutations) in the AMOVA, indicating negligible or no gene flow between the clades identified by the phylogenetic and population genetic analyses. In contrast, Φ SC, which was also significant (P=0.000), was very low (0.041), suggesting unrestricted gene flow between colour morphs and locations within each of the clades (Table 2).
To further investigate population structure between colour morphs and geographic locations, individual comparisons of the “among population” component of genetic variation between different groups were carried out. Results supported strong and significant population structure between KB and the GBR morphs, between KB and the pink morph from Loloata, as well as between the GBR morphs and the pink morph from Loloata (Φ ST=0.905–0.979) (Table 3). Φ ST values for all other within clade comparisons (e.g. LI versus OT, yellow versus brown) were relatively low, but significant (Φ ST=0.033–0.248) (Table 3). The Φ ST values greater than 0.2 for GBR–PNG groups indicate that there is more restricted gene flow between the GBR and PNG locations than along the entire GBR (LI to OTI, Φ ST=0.066), which shows the highest levels of gene flow measured, though there is some genetic structure at this spatial scale along the GBR for P. fuscus (see Hartl 2000).
Molecular clock
The relative rates test indicated that the operation of a molecular clock could not be rejected (P>0.05). A molecular clock was therefore enforced to estimate ages of the three lineages. An average divergence rate of 6.41% MY−1 was calculated based on the 192 bp of the hypervariable region evolving at 12.9% MY−1 and the 257 bp of the conserved region evolving at 1.1% MY−1 from the sequenced dloop fragment, as per Alvarado et al. (1995). The estimated divergence rate of 6.41% MY−1 is similar to the divergence rate found for cichlids using a similar segment of the mitochondrial control region (6.5–8.8%). It is faster than other rates suggested for dloop of fish in the literature (1.5 (slow) – 3.6%), but most of these studies investigated the entire control region (Martin and Palumbi 1993; Cantatore et al. 1994; Stepien and Faber 1998; Donaldson and Wilson 1999; Bowen et al. 2001; Waters and Wallis 2001). Based on a divergence rate of 6.41% MY−1, the Kimbe Bay clade and Loloata Island pink morph diverged from their common ancestor approximately 2.4 million years ago (Ma). Time since divergence of the Kimbe Bay clade from the common ancestor to the GBR and southern PNG clade would have been approximately 4.0 Ma.
Morphology
The morphometric analysis supported the separation of the Loloata Island pink morph from all other morphs. Nearly all individuals of the Loloata Island pink morph had 27 dorsal rays, whereas the other morphs mostly had a range of 25 to 27 dorsal fin rays (Table 4). The different colour morphs from Kimbe Bay and the yellow and brown morphs from Loloata Island had similar frequencies of 25, 26 and 27 rays. In contrast, the yellow and brown morphs from Lizard Island mostly had 26 dorsal fin rays. The brown and yellow morphs from One Tree Island had similar frequencies of 25 and 26 rays.
Discussion
The phylogenetic analysis and haplotype network clearly identified three distinct clades of Pseudochromis fuscus that relate to a combination of colour morphs and geographic location. The first clade included yellow and brown morphs from three geographic locations, One Tree Island, Lizard Island and Loloata Island, as well as the orange morph from Loloata Island. The second clade contained all three colour morphs from Kimbe Bay (pink, orange and grey). Thirdly, the pink morph from Loloata Island formed a distinct clade. Morphological data supported the distinction of the Loloata Island pink morph. The extremely high Φ ST values (0.905–0.979) signify that gene flow between the populations represented by the three clades is negligible. Furthermore, the indices of haplotype (h=0.96) and nucleotide (π=12.8%) diversity indicate deep divergences among haplotypes from the different clades. In combination, these results generate three important conclusions: (1) the yellow, brown, and orange morphs found on the Great Barrier Reef and in southern Papua New Guinea are likely to represent a single polychromatic species, (2) there is a strong genetic break between populations of P. fuscus in northern PNG and those in southern PNG and GBR, and (3) the pink morph of P. fuscus from southern PNG is genetically distinct and may represent a separate species.
Colour had a relatively poor relationship with genetic structure in P. fuscus. Although the pink morph from Loloata Island formed a distinct clade, the yellow and brown morphs from three locations (OT, LI, LOL), as well as the orange morph from Loloata Island, were represented in a single clade. All three morphs from Kimbe Bay also belonged to a single clade. The pink and orange morphs from Kimbe Bay are sex-limited and thought to be males and females of the same species (Eyre 2001). Therefore, the absence of genetic differentiation between these morphs was expected. The status of the grey morph in Kimbe Bay is less clear. Colour morphs in Kimbe Bay can often be difficult to distinguish and the grey morph might simply represent a darker form of either the pink or orange morphs.
In contrast to the different colour morphs in Kimbe Bay, the yellow and brown morphs from the GBR and southern PNG are not sex-limited (Munday et al. 2003). Nevertheless, molecular analyses have found no genetic differentiation between these morphs (this study; Messmer et al. 2005) and experiments have revealed the potential for colour change from yellow to brown under appropriate environmental conditions (Gill 2004; Messmer et al. 2005). Consequently, the brown and yellow morphs of P. fuscus on the GBR and in southern PNG, along with the orange morph in southern PNG, are almost certainly colour variants of the same species. In all cases lacking genetic differentiation, insufficient time may have elapsed since isolation for lineages to become reciprocally monophyletic, particularly if effective population sizes are large, as is the case for many coral reef fishes which may have diverged very recently (Lacson 1994; Miya and Nishida 1997; McMillan et al. 1999; Knowlton 2000). However, if this were the case, we would expect to see some structure within clades containing a mixture of morphs. Furthermore, the capacity for colour change, at least from yellow to brown, indicates that this is a single polymorphic species and we would not expect to see genetic differentiation between the colour morphs.
Although the pink morph from southern PNG was genetically and morphologically distinct, 15 individuals of this morph that were histologically examined were all found to be female (Messmer, unpublished data). It is unclear at this stage if males of the pink morph were not sampled or if there is a differently coloured male morph that was not collected. Nevertheless, differences in body colouration between the pink morphs from Loloata and Kimbe Bay in addition to the strong genetic divergence suggest that these two pink morphs are distinct and reproductively isolated.
Population genetic structure of P. fuscus was associated with geographic location, and to a lesser degree colour (above), but not distance. One Tree Island, Lizard Island and Loloata Island are separated by approximately 1,650 km and were represented in a single clade. In contrast, the distance between Kimbe Bay and Loloata Island is only about 550 km, yet populations from these two locations were genetically distinct (d=15.5%). Furthermore, gene flow was more extensive between One Tree Island and Lizard Island than between Lizard Island and Loloata Island, even though the former pair of locations is further apart than the latter pair. Finally, the pink morph from Loloata Island was found to be genetically distinct from the other three morphs (yellow, brown and orange) occurring in the same geographic location. All these results demonstrate that isolation by distance does not explain the observed genetic structure between geographic locations in the P. fuscus complex.
Barriers to dispersal and patterns of ocean circulation appear to explain genetic differentiation between populations of some marine species (eg. Bohonak 1999; Nelson et al. 2000; Muss et al. 2001) and could help explain the genetic structure we detected among populations of P. fuscus. Kimbe Bay and Loloata Island are separated by two substantial landmasses (the eastern peninsula of Papua New Guinea and the Island of New Britain) that could present a direct physical barrier to the movement of larval and adult coral reef fishes. These landmasses also mean that the circulation patterns in the Gulf of Papua and the east Bismark Sea are not strongly connected. The Gulf of Papua (where Loloata Island is situated, Fig. 1) has complex patterns of water circulation, including gyres that could retain dispersive propagules (Woolfe et al. 1997). Although a deep strait of over 1,000 m depth separates mainland New Guinea and New Britain (Fig. 1) and would have remained open during periods of low sea level, currents moving through this strait tend to flow northwest along the coast of PNG (Lindstrom et al. 1987; Pinkard et al. 1997) and do not appear to be strongly connected with the southeast Bismark Sea (where Kimbe Bay is located). Consequently, restricted gene flow as a result of geography may have played a role in the deep genetic divergence seen between morphs of P. fuscus at Loloata Island and Kimbe Bay.
In contrast, the absence of barriers to dispersal and the presence of currents favourable to transport between locations probably explain the gene flow between populations of P. fuscus on the Great Barrier Reef. The East Australian Current provides a strong oceanographic link between the northern and southern GBR (Anonymous 2000). Furthermore, the GBR is effectively a continuous band of suitable habitat, which can aid dispersal and help maintain strong genetic connectivity between populations (Riginos and Nachman 2001; Rocha et al. 2002; Rocha 2004).
Factors favouring gene flow between the GBR and southern PNG are likely to be intermediate to the scenarios described above. There is no geographic barrier to dispersal between the northern GBR and southern PNG. However, water circulation in this area is complex and variable (Lindstrom et al. 1987; Pinkard et al. 1997; Woolfe et al. 1997). Also, areas of reef that may act as stepping stones to dispersal are not continuous in this region. In particular, there is a significant break in the distribution of coral reefs where the Fly, Kikori, and other large rivers flow into the Gulf of Papua (Wolanski et al. 1984). These rivers discharge large volumes of freshwater, with a long residence time in the Gulf of Papua (Wolanski et al. 1984). Unfavourable conditions for the growth of coral reefs limits the habitat available for adult P. fuscus in this area and may also provide a soft barrier to dispersal of larval fishes. Genetic breaks associated with the discharge of large rivers have been reported in other locations (Rocha et al. 2002; Rocha 2004).
Population structures of each clade in P. fuscus showed high haplotype diversities, but variable nucleotide diversities, suggesting that each clade may have a unique evolutionary history. It appears that the GBR clade went through a bottleneck most recently (low π, 0.57%), followed by a rapid expansion (high h, 0.92). This is consistent with glacial cycles over the past 1.8 MA that have repeatedly left the shallow Great Barrier Reef lagoon uninhabitable for coral reef organisms. Expansion of relictual Great Barrier Reef P. fuscus populations into the GBR lagoon, following periods of low sea level, could explain the genetic bottleneck and evidence of rapid population expansion. Similar genetic evidence for reinvasion and population expansion on the GBR is seen in A. polyacanthus (van Herwerden and Doherty 2005).
The Kimbe Bay clade has had a longer evolutionary history (high π, 2.98%; high h, 0.92), which is consistent with the tectonic history of New Britain and the presence of deep-water reefs in close proximity (a few kilometres) to the New Britain mainland. Many of the coral reefs in this area rise to the surface from over 200 m depth (Holthus 1994) and could have been refuges for reef fishes during periods of low sea level. Time since divergence of the PNG and GBR P. fuscus lineages (~2.4–4 Ma) was estimated to pre-date the most recent glacial cycles which have driven major fluctuations in sea levels during the last 1.8 Ma (Becquey and Gersonde 2002; Lambeck et al. 2002), indicating that recent sea level changes have been less important in the evolutionary history of P. fuscus in Kimbe Bay than they have on the GBR.
New Britain is tectonically different from the majority of the mainland of New Guinea, which is part of the Australian plate (Hall 1998). Over the last 40 Ma, the Australian shelf, which was previously positioned much closer to Antarctica, gradually moved north. The northern parts of New Guinea, including New Britain, originated from several island arcs, which were formed in the north eastern Pacific (Hall 1998). Over the last 40 Ma, these island arcs moved to the south west and some of them collided with the New Guinea mainland. Others, such as New Britain, are still islands lying to the north east of the New Guinea mainland (Hall 1998; Polhemus and Polhemus 1998). Different biogeographic origins of the Kimbe Bay and Loloata Island populations in the past may have contributed to the observed divergence between these populations.
The deep divergence between the three groups identified here strongly suggests that the current taxonomy of P. fuscus should be reconsidered and that each clade is likely to represent a distinct species. Criteria for the recognition of species by molecular evidence are inconsistent. However, P. fuscus meets several criteria that have been used to assign species status. Levels of genetic differentiation (0.905–0.979) between the different clades are similar to that used to assign species status in other reef fishes that exhibit subtle differences in colouration (e.g. Rocha 2004). The orange and pink morphs from KB are sex-limited (Eyre 2001), whereas the yellow and brown morphs from the GBR appear to have an ecological basis (Munday et al. 2003), therefore, these two groups are biologically and ecologically quite different. Furthermore, the two populations differ in colouration as well as dorsal fin ray counts, adding a third component to the differentiation between colour variants. Additional information is required for the pink morph from Loloata before the species status of this variant can be properly evaluated. If both sexes are found within the pink morph from Loloata, then it is likely to represent a different species to the pink morph from Kimbe Bay, given the genetic divergence, differences in colouration and the biogeographic history of the two locations. Mitochondrial DNA is almost exclusively transmitted through the maternal lineage and may identify different relationships to those identified by nuclear data (e.g. Lu et al. 2001). Ideally, nuclear markers will now be used to further assess genetic interactions between colour morphs and locations in P. fuscus.
References
Alvarado BJR, Baker AJ and Mejuto J (1995) Mitochondrial DNA control region sequences indicate extensive mixing of swordfish (Xiphias gladius) populations in the Atlantic Ocean. Can J Fish Aquat Sci 52:1720–1732
Anonymous (2000). The East Australian Current. CSIRO Marine Research Information sheet 37, CSIRO, Australia
Avise JC (1998) The history and purview of phylogeography: a personal reflection. Mol Ecol 7(4):371–379
Avise JC (2000) Phylogeography: the history and formation of species. Harvard University Press, Cambridge MA
Becquey S, Gersonde R (2002) Past hydrographic and climatic changes in the Subantarctic Zone of the South Atlantic—the Pleistocene record from ODP Site 1090. Paleogeogr Paleoclimatol Paleoecol 182:221–239
Bellwood DR, Wainwright PC (2002) The history and biogeography of fishes on coral reefs. In: Sale PF (ed) Coral reef fishes: dynamics and diversity in a complex ecosystem. Academic, Amsterdam, pp 5–32
Bernardi G, Holbrook SJ, Schmitt RJ, Crane NL, DeMartini E (2002) Species boundaries, populations and colour morphs in the coral reef three-spot damselfish (Dascyllus trimaculatus) species complex. Proc Roy Soc Lond B 269:599–605
Bohonak AJ (1999) Dispersal, gene flow, and population structure. Quart Rev Biol 74:21–45
Bowen BW, Bass AL, Rocha LA, Grant WS, Robertson DR (2001) Phylogeography of the trumpetfishes (Aulostomus): ring species complex on a global scale. Evolution 55:1029–1039
Cantatore P, Roberti M, Pesole G, Ludovico A, Milella F, Gadaleta MN, Saccone C (1994) Evolutionary analysis of cytochrome b sequences in some perciformes—evidence for a slower rate of evolution than in mammals. J Mol Evol 39:589–597
Colborn J, Crabtree RE, Shaklee JB, Pfeiler E, Bowen BW (2001) The evolutionary enigma of bonefishes (Albula spp.): cryptic species and ancient separations in a globally distributed shorefish. Evolution 55:807–820
DeMartini EE, Donaldson TJ (1996) Color morph-habitat relations in the arc-eye hawkfish Paracirrhites arcatus (Pisces: Cirrhitidae). Copeia 1996:362–371
Doherty PJ, Planes S, Mather P (1995) Gene flow and larval duration in seven species of fish from the Great Barrier Reef. Ecology 76:2373–2391
Domier ML (1994) Speciation in the serranid fish Hypoplectrus. Bull Mar Sci 54: 103–141
Donaldson KA, Wilson RR Jr. (1999) Amphi-panamic geminates of snook (Percoidei: Centropomidae) provide a calibration of the divergence rate in the mitochondrial DNA control region of fishes. Mol Phylog Evol 13:208–213
Excoffier L, Smouse P, Quattro JM (1992) Analysis of molecular variance inferred from metric distance among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:343–359
Eyre PJ (2001) Colour, gender and sex allocation in fishes of the family Pseudochromidae. Honours Thesis. School of Marine Biology and Aquaculture, James Cook University, Townsville
Fischer EA (1980) Speciation in the Hamlets (Hypoplectrus: Serranidae)—a continuing enigma. Copeia 4:649–659
Florence WK, Hulley RA, Stewart BA, Gibbons MJ (2002) Genetic and morphological variation of the lanternfish Lampanyctodes hectoris (Myctophiformes : Myctophidae) off southern Africa. South African J Mar Sci 24:193–203
Garcia-Machado E, Chevalier Monteagudo PP, Solignac M (2004) Lack of mtDNA differentiation among hamlets (Hypoplectrus, Serranidae). Mar Biol 144:147–152
Gill AC (2004) Revision of the Indo-Pacific dottyback fish subfamily Pseudochrominae (Perciformes: Pseudochromidae). Smithiana Monographs 1:1–214
Hall R (1998) The plate tectonics of Cenozoic SE Asia and the distribution of land and sea. In: Holloway JD (ed) Biogeography and geological evolution of SE Asia. Backhuys Publishers, Leiden, pp 99–131
Hartl DL (2000) A primer of population genetics. Sinauer Associates Inc., Sunderland, pp 68–72
Holthus P (1994) Kimbe Bay rapid ecological assessment: the coral reefs of Kimbe Bay. Vol.1: synthesis report. The Nature Conservancy, Auckland
Knowlton N (2000) Molecular genetic analyses of species boundaries in the sea. Hydrobiologia 420:73–90
Kumar S, Tamura K, Jakobsen IB, Nei M (2001) MEGA2: molecular evolutionary genetics analysis software. Bioinformatics 17:1244–1245
Lacson JM (1994) Fixed allele frequency differences among Palauan and Okinawan populations of the damselfishes Chrysiptera cyanea and Pomacentrus coelestis. Mar Biol 118:359–365
Lambeck K, Yokoyama Y, Purcell T (2002) Into and out of the last glacial maximum: sea-level changes during oxygen isotope stages 3 and 2. Quaternary Sci Rev 21:343–360
Lee WJ, Conroy J, Howell WH, Kocher TD (1995) Structure and evolution of teleost mitochondrial control regions. J Mol Evol 41:54–66
Lieske E, Meyers R (1999) Coral reef fishes. Princeton University Press, Princeton
Lindstrom E, Lukas R, Fine R, Firing E, Godfrey S, Meyers G, Tsuchiya M (1987) The Western Equatorial Pacific Ocean Circulation study. Nature 330:533–537
Lu G, Basley DJ, Bernatchez L (2001) Contrasting patterns of mitochondrial and microsatellite introgressive hybridisation between lineages of lake whitefish (Coregonus clupearformis); relevance for speciation. Mol Ecol 10: 965–985
Martin AP, Palumbi SR (1993) Body size, metabolic rate, generation time, and the molecular clock. Proc Nat Acad Sci USA 90:4087–4091
McMillan WO, Palumbi SR (1997) Rapid rate of control-region evolution in Pacific butterflyfishes (Chaetodontidae). J Mol Evol 45:473–484
McMillan WO, Weigt LA, Palumbi SR (1999) Color pattern evolution, assortative mating, and genetic differentiation in brightly colored butterflyfishes (Chaetodontidae). Evolution 53:247–260
Messmer V (2003) Genetic and experimental analysis of colour variation in coral reef fish (family: Pseudochromidae). Honours Thesis. School of Marine Biology and Aquaculture, James Cook University, Townsville
Messmer V, Jones GP, van Herwerden L, Munday PL (2005) Genetic and ecological characterisation of colour dimorphism in a coral reef fish. Env Biol Fish (in press)
Michael SW (2004) Basslets, Dottybacks and Hawkfishes. T.F.H. Publications, Neptune City
Miya M, Nishida M (1997) Speciation in the open ocean. Nature 389:803–804
Munday PL, Eyre PJ, Jones GP (2003) Ecological mechanisms for coexistence of colour polymorphism in a coral reef fish: an experimental evaluation. Oecologia 137:519–526
Muss A, Robertson DR, Stepien CA, Wirtz P, Bowen BW (2001) Phylogeography of Ophioblennius: the role of ocean currents and geography in reef fish evolution. Evolution 55:561–572
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Nei M, Kumar S (2000) Molecular evolution and phylogenetics. Oxford University Press, Oxford
Nelson JS, Hoddell RJ, Chou LM, Chan WK, Phang VPE (2000) Phylogeographic structure of false clownfish, Amphiprion ocellaris, explained by sea level changes on the Sunda shelf. Mar Biol 137:727–736
Orbacz EA, Gaffney PM (2000) Genetic structure of tautog (Tautoga onitis) populations assayed by RFLP and DGGE analysis of mitochondrial and nuclear genes. Fish Bull 98:336–344
Palumbi SR (1994) Genetic divergence, reproductive isolation, and marine speciation. Ann Rev Ecol Syst 25:547–572
Palumbi SR (1997) Molecular biogeography of the pacific. Coral Reefs 16(Suppl): 52
Pickard GL, Donguy JR, Henin C, Rougerie F (1977) A review of the physical oceanography of the Great Barrier Reef and Western Coral Sea: Circulation. Aust Inst Mar Sci Monogr Ser 2:109–121
Planes S (2002) Biogeography and larval dispersal inferred from population genetic analysis. In: Sale PF (ed) Coral reef fishes: dynamics and diversity in a complex ecosystem. Academic, Amsterdam, pp 201–220
Planes S, Doherty PJ (1997) Genetic and color interactions at a contact zone of Acanthochromis polyacanthus—a marine fish lacking pelagic larvae. Evolution 51:1232–1243
Planes S, Fauvelot C (2002). Isolation by distance and vicariance drive genetic structure of a coral reef fish in the Pacific Ocean. Evolution 56:378–399
Polhemus DA, Polhemus JT (1998) Assembling New Guinea: 40 million years of island arc accretion as indicated by the distribution of aquatic Heteroptera (Insecta). In Holloway JD (ed) Biogeography and geological evolution of SE Asia. Backhuys Publishers, Leiden, pp 327–340
Posada D, Crandall KA (1998) Modeltest: testing the model of DNA substitution. Bioinformatics 14:817–818
Rambaut A (2003) Se-Al, a manual sequence alignment editor
Ramon ML, Lobel PS, Sorenson MD (2003) Lack of mitochondrial genetic structure in hamlets (Hypoplectrus spp.); recent speciation or ongoing hybridization. Mol Ecol 12: 2975–2980
Raymond M, Rousset F (1995). An exact test for population differentiation. Evolution 49:1280–1283
Riginos C, Nachman MW (2001) Population subdivision in marine environments: The contributions of biogeography, geographical distance and discontinuous habitat to genetic differentiation in a blennioid fish, Axoclinus nigricaudus. Mol Ecol 10:1439–1453
Rocha LA (2004) Mitochondrial DNA and colour pattern variation in three western Atlantic Halichoeres (Labridae), with the revalidation of two species. Copeia 1994(4):770–782
Rocha LA, Bass AL, Robertson DR, Bowen BW (2002) Adult habitat preference, larval dispersal, and the comparative phylogeography of three Atlantic surgeonfishes (Teleostei: Acanthuridae). Mol Ecol 11:243–252
Rohlf FJ (1973) Algorithm 76. Hierarchical clustering using the minimum spanning tree. Comput J 16:93–95
Sambrook J, Fritch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbour Laboratory Press, Cold Spring Harbour
Schneider S, Roessli D, Excoffier L (2000) Arlequin: a software for population genetics data analysis. Ver 2.000. Genetics and Biometry Lab, Deptartment of Anthropology, University of Geneva, Geneva
Stepien CA, Faber JE (1998) Population genetic structure, phylogeography and spawning philopatry in walleye (Stizostedion vitreum) from mitochondrial DNA control region sequences. Mol Ecol 7:1757–1769
Sturmbauer C, Baric S, Salzburger W, Ruber L, Verheyen E (2001) Lake level fluctuations synchronize genetic divergences of cichlid fishes in African lakes. Mol Biol Evol 18:144–154
Swofford DL (1998) PAUP* phylogenetic analysis using parsimony (* and other methods). Sinauer Associates, Sunderland
Taylor MS, Hellberg ME (2002) Genetic evidence for local retention of pelagic larvae in a Caribbean Reef Fish. Science 299:107–109
Thacker CE (2004) Population structure in two species of the coral reef goby Gnatholepis (Teleostei: Perciformes) among four South Pacific island groups. Coral Reefs 23:357–366
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG (1997). The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 24:4876–4882
Thresher RE (1978). Polymorphism, mimicry, and the evolution of the hamlets (Hypoplectrus, Serranidae). Bull Mar Sci 28:345–353
van Herwerden L, Doherty PJ (2005) Contrasting patterns of mitochondrial introgression between color morphs of a tropical reef fish (Acanthochromis polyacanthus) at two zones of secondary contact on the Great Barrier Reef. J Evol Biol. DOI 10.1111/j.1420-9101.2005.00969.x
Ward RD, Woodwark M, Skibinski DOF (1994) A comparison of genetic diversity levels in marine, freshwater, and anadromous fishes. J Fish Biol 44:213–232
Waters JM, Wallis GP (2001) Mitochondrial DNA phylogenetics of the Galaxias vulgaris complex from South Island, New Zealand: rapid radiation of a species flock. J Fish Biol 58:1166–1180
Wolanski E, Pinckard GL, Jupp DLB (1984) River plumes, coral reefs and mixing in the Gulf of Papua and the nrothern Great Barrier Reef. Est, Coast Shelf Sci 18:291–314
Woolfe KJ, Larcombe P, Whitmore GP (1997) Marine geology of the Western Gulf of Papua. James Cook University of North Queensland, Townsville
Acknowledgements
We thank Dik Knight of Loloata Island Resort and Max Benjamin of Walindi Plantation Resort for support of our research in PNG. Mahonia Na Dari Research and Conservation Centre provided excellent logistic support. Tony Gill generously shared his knowledge of pseudochromid taxonomy and biology and Lance Bode provided helpful information on oceanographic models of the region. This research was supported by James Cook University grants to LVH and Australian Research Council grants to GPJ and PLM.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Biological Editor R.C. Carpenter
Rights and permissions
About this article
Cite this article
Messmer, V., van Herwerden, L., Munday, P.L. et al. Phylogeography of colour polymorphism in the coral reef fish Pseudochromis fuscus, from Papua New Guinea and the Great Barrier Reef. Coral Reefs 24, 392–402 (2005). https://doi.org/10.1007/s00338-005-0001-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00338-005-0001-9