
Abstract
Interferon-stimulated genes (ISGs) are the effectors of interferon (IFN) actions and play major roles in innate immune defense against microbial infection. During virus infection, ISGs impart antiviral actions to control virus replication and spread but can also contribute to disease pathology if their expression is unchecked. Antiviral ISGs have been identified by a variety of biochemical, genetic, and virologic methods. New computational approaches are expanding and redefining ISGs as responders to a variety of stimuli beyond IFNs, including virus infection, stress, and other events that induce cytokines. These studies reveal that the expression of ISG subsets link to interferon regulatory factors (IRF)s, NF-kB, and other transcription factors that impart gene expression in specific cell types independently of IFNs, including stem cells and other cell types where ISGs are constitutively expressed. Here, we provide a broad overview of ISGs, define virus-induced genes (VSG)s, and discuss the application of computational approaches and bioinformatics platforms to evaluate the functional role of ISGs in epigenetics, immune programming, and vaccine responses.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
IFN was first characterized over 50 years ago as a soluble factor induced by influenza A virus infection (Isaacs et al. 2015; Probst et al. 2017). Since then ISGs have been defined as those genes whose expression are induced or regulated by IFN, including types I–III IFNs (Der et al. 1998; Chow and Gale 2015). The classification of IFNs as types I, II, or III were made according to their order of discovery. Each uses unique receptors; all IFNs signal through the JAK-STAT pathway, but each receptor type utilizes discrete and overlapping signaling factors to similarly produce intersecting, yet distinct cell and tissue type specific ISG profiles (Chow and Gale 2015; Henle 1950; Schoggins et al. 2014; Schneider et al. 2014). Today, over 500 ISGs have been defined as genes with differential gene expression in response to IFNs (de Veer et al. 2001). Advances in biotechnology and bioinformatics are allowing us to expand our definition of ISGs to better understand their role in infection and immunity. Below we explore the current understanding of ISG regulation, and we discuss how bioinformatics approaches can be applied to expand our knowledge of ISGs and their function.
IFN induction by host innate immune responses
During virus infection, IFN is first induced intrinsically in infected cells through a process of host cell recognition of viral RNA products, or other viral macromolecules called pathogen-associated molecular patterns (PAMPs). PAMPs are physically recognized as non-self by binding to specific cellular pathogen recognition receptors (PRRs) such as Toll-like receptors (TLRs), RIG-I-like receptors (RLRs), NOD-like receptors (Chow and Gale 2015). PAMP binding induces PRR-linked signaling cascades that drive antiviral activity through activation of latent transcription factors, interferon regulator factors (IRF)s, NF-κB, and others to induce target gene expression. In various cell types, NF-κB and IRFs drive the expression and secretion of Type I and III IFNs from the site of infection (Hertzog et al. 2011; Dixit et al. 2010) (Fig. 1). Secreted IFN then engages the specific IFN receptor on the infected cell and neighboring bystander cells within the local tissue to induce ISG expression. Moreover, upon PAMP stimulation of specialized IFN-producing plasmacytoid dendritic cells, IFN is produced systemically in the circulation (Webster et al. 2016). Ideally, ISG induction is a transient event wherein ISG actions suppress virus infection and protect cells and tissue from disease. Upon resolution of virus infection and subsequent PAMP suppression, typically the IFN response is shut off by the expression of a second set of ISGs that negatively regulate innate immune actions to resolve the IFN response (Schoggins et al. 2014). However, IFNs can be toxic if the response is not resolved appropriately. Indeed, constitutive IFN production and response are linked with a variety of autoimmune diseases labeled as interferonopathies (Crow 2016; Rodero and Crow 2016). Thus, ISG induction and resolution are equally essential to avoid pathology from virus infection and other microbial challenges, and better understanding of these regulatory pathways will be essential to developing clinical applications that can improve host resistance to infection and healthy recovery from infection.

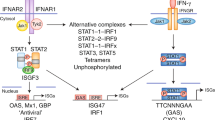
Interferon signaling. Each of the three interferon types (Type I–III) is routed based on their receptor complex. Additional information can be found in the text
Similar yet distinct IFN signaling pathways
Interferons are named by their order of discovery and are further characterized by their genomic location and homology to define three classes: type I, type II, and type III (Borden et al. 2007) (see Fig. 1). Type I interferons, which include IFN-α and β and other minor subtypes (Stark and Darnell 2012; Henle 1950; Chow and Gale 2015), bind the type I IFN receptor through two subunits, IFNR1 and IFNR2. IFNR1 and IFNR2 are expressed on most cells of the body, though recent studies indicate that cells in the gut express limited type I IFN receptor (Pott et al. 2017). Type II IFN, also known as IFN-γ, binds to IFNGR1 and IFNGR2 receptors present on most cell types, and serves to polarize the immune response toward an inflammatory and antiviral/antimicrobial phenotype (Schroder et al. 2004). Type II interferons induce receptor-mediated JAK1 and JAK2 phosphorylation to activate STAT1 by the formation of STAT1 homodimers. Dimerized STAT1 translocates from the cellular cytoplasm to the nucleus, binds the gamma activator sequence (GAS) on IFN-γ-inducible genes and activates their transcription. Finally, type III IFN comprises a four-gene family in humans, IFNL1, INFL2, IFNL3 (also known as IL29, IL28A, and IL28B), and IFNL4 either generating a mature protein or a nonfunctional frameshift variant (Hemann et al. 2017). Their corresponding proteins are IFNl1, INFl2, IFNl3, IFNl4. The type III IFN receptor comprises IFNλR1 and IL-10R2 subunits. Both type I and type III IFN receptor signaling activate the JAK-STAT pathway through receptor phosphorylation directed by receptor-bound JAK1 (Janus kinase I) and TYK2 (Tyrosine kinase 2) members of the JAK kinase family. The phosphorylation events directed by these kinases recruit and activate signal transducer and activator of transcription STAT1 and STAT2 proteins, leading to their interaction with IRF9 to form the ISGF3 (Interferon-stimulated effector factor 3) complex that moves into the nucleus to trigger ISG expression by binding to the interferon-stimulated response element (ISRE) on target promoters. These mechanisms are well defined and have been reviewed for their role in host defense (Schoggins et al. 2011; Raftery and Stevenson 2017).
A subset of ISGs are often induced within the infected cell during acute virus infection even without the induction IFN and IFN signaling. These genes are induced by virus infection through PAMP/PRR signaling pathways that activate IRFs, and other transcription factors, by IRF binding sites within their promotor regions that encode ISRE or GAS elements to confer induction by IFN. These classes of ISGs are true first responders to virus infection and can be called virus stimulated genes (VSGs) (Henle 1950; Schoggins et al. 2014; Schneider et al. 2014). Many VSGs were defined in studies of virus infection in specific gene knockout mice and from human genetics studies (Schoggins et al. 2014; Schneider et al. 2014) and summarized in Fig. 2. Importantly, new bioinformatics/computational approaches of virus-response gene sets are extending our understanding of VSGs (Fig. 3).

VSG network. A network of viral-stimulated genes (VSG) and their viral triggers. The nodes represent a VSG (pink) or a virus (green). Each hubs’ size indicates its level of connectivity. The network also illustrates which VSGs are unique and which ones are common hubs

An overview of strategies and platforms in ISG discovery. A break down of conventional, emerging, and future methods to deciphering ISGs
The IRF3 transcription factor represents the major node for transcription of VSGs induced by PRR signaling as direct IRF3-target genes. In a pivotal study, Hiscott and colleagues (Grandvaux et al. 2002) revealed the set of IRF3-target genes induced or regulated by the inducible expression of a constitutively activated version of IRF3 in cultured cells. This study identified genes that were induced (up-regulated) or suppressed (down-regulated) upon IRF3 activation (Table 1). Several of these target genes are VSGs known for their roles in antiviral immunity (for example IFIT1, IFIT2, IFIT3, RSAD2, ISG15, and SAMHD1) (Schoggins et al. 2014). Previous studies (Sarkar and Sen 2004) observed genes activated by double-stranded RNA and other viral products and noted their cellular pressures as viral stress-inducible genes (VSIG).
Tools to distinguish VSGs and ISGs into functional categories
Bioinformatic approaches categorizing genes into functional classes have expanded in recent years (Fig. 4). Traditionally, genes are grouped and scored by their representation and connections with biological functions in public data bases. These approaches include gene ontology (GO) terms, (KEGG), Gene Set Enrichment (GSEA), Search Tool for the Retrieval of Interacting Genes/Proteins (STRING), BIOGRID, Ingenuity Pathway Analysis (IPA), The Database for Annotation, Visualization and Integrated Discovery (DAVID), and other databases and computational tools. A common analytical process is to define genes by over-representation analysis (ORA) within known functional categories that count how many common genes are represented within a defined set and calculate a statistical cut off or overlap. GSEA (Hung et al. 2012) operates in a similar manner but without the statistical cutoff, ranking all the genes in the genome and testing whether a user’s input genes are over-represented compared to all genes. GSEA is powerful, but it is highly sensitive to non-categorical gene “noise” and data may not always be applicable toward defining gene function (Liu et al. 2017). These functional tools (GSEA, GO, etc.) can be underpowered in the detection of ISGs because their descriptive outputs like “response to virus” and “interferon signaling” do not distinguish between the distinct interferon driven and antiviral mechanisms driving ISGs activation. Additional approaches such as gene co-expression analysis, gene network analysis, and protein–protein interaction network analysis consider the concept of shared behavior between genes that can also identify gene response networks driven by VSGs and ISGs.

An overview of tools for classification and functional analysis
Using databases to define VSG and ISG function
Functional tools can vary in how they group genes to function, including using computational-derived gene signatures (wherein a literature-mining algorithm associates gene with function), as well as application of manually curated data bases (in which a human reads a manuscript and documents the findings of gene function into said database). Over the years, the largest contribution to these approaches has been the increase in publically available data sets. Beyond GSEA, alternate approaches leveraging publicly available data provide greater statistic power while identifying information on new genes and transcriptional regulators. Recent work by (Shaw et al. 2017) focused on cross species response to type I interferons. The authors identified unique ancestral including genes not previously associated with IFN and produced a database to query expression signatures. Broader efforts to categorize VSGs and ISGs through publicly available data are underway. Below we feature a few databases that are increasingly useful in defining VSG/ISG function.
Interferome (http://www.interferome.org) is a large curated database of interferon-stimulated genes produced from published microarray experiments (Samarajiwa et al. 2009; Hertzog et al. 2011; Rusinova et al. 2013). This database has catalogued over six thousand ISGs in human studies and over five thousand ISGs in mouse studies. The system also has curated genes according to interferon type, noting 3553 type I ISGs in human and 3704 in mice. The web portal allows users to query their own gene signatures against an IFN database. This is a valuable resource for researchers in mining public microarray data. Interferome can summarize ISGs that share common transcriptional regulators like IRF and NF-κB. One limitation is that Interferome’s database is built from microarray data and has not yet been updated with sequencing data. In contrast, the ARCHS4 database was recently published in an attempt to leverage all RNA-seq and ChIP-seq data (Lachmann et al. 2018). This approach uses all the human and mouse genomics sequencing data from public repositories SRA (sequence read archive) and GEO (gene expression omnibus) and provides web-based analysis and visualization tools. In ARCHS4, the user can search for their genes of interest within RNAseq datasets. The results are derived from genes and pathways that share co-expression patterns. Downstream analytics tools provide predicted biological functions, transcription factors, protein–protein interactions, and expression within tissue types. As an alternative approach, users can also perform metadata searches and simplistically download the raw data from SRA and GEO. Overall, ARCHS4 houses 137,792 samples (72,363 mice and 65,429 human) along with simplistic web tools.
Interferome and ARCHS4 are great resources with very broad capabilities; however, other researchers have built tools with a focus on the unique expression signatures found in immune cells. The Immunological Genome Project (http://www.Immgen.org) is a public resource of expression data disseminated from mouse immune cells, and has been a valuable resource for immunologists and computational biologists, providing insights into cell differentiation and immune regulatory pathways. Utilizing Immgen’s database, a group of researchers (Mostafavi et al. 2016) identified ISG modules and built a regulatory network they tested across species (human and mouse) using external data sets. After identifying gene modules, Mostafavi et al. used ATACseq to verify the start sites during ISG induction. ATAC-seq (Assay for Transposase-Accessible Chromatin using sequencing) is an approach that uncovers chromatin accessible regions. Their analysis identified regulatory genes by known factors (STAT 1/2, IRF9, ISRE) and other non-canonical and not STAT-dependent ones. This study is an excellent example of combining previous techniques (microarray, data from Immgen) and new approaches (data mining, co-expression analysis, ATACseq). Their results distinguished ISGs by kinetics and cell-type specificity, while also targeting ISGs specific to JAK inhibition. Public data efforts like Interferome and Immgen allow users to cross reference their findings with published experimental data, but these approaches have not adopted newer sequence-based approaches.
Emerging technologies for ISG discovery
Technologies like RNAseq, CHIPseq, ATACseq, and Clipseq have expanded our understanding of viral-induced ISGs and produce results faster and cheaper than previous methods. RNAseq has the advantage of studying viral sequences and host VSG expression synergistically. As new annotation and reference sequences become available, data can be realigned and reanalyzed. This approach is different from earlier microarray approaches where reference sequences were fixed onto arrays. If a reference sequence is not available then approaches like de novo assembly (Sohn and Nam 2018) can identify a consensus reference sequence.
RNAseq broadens the genomic landscape by observing more than just gene expression; it also allows one to observe differential isoforms, non-coding, and un-annotated regions not seen in previous platforms. An example of this type of discovery is the use of single cell sequencing (SC-seq) which revealed a non-IFN dependent role of IFNB1 in early viral recognition (Doganay et al. 2017). In this study, researchers applied a single cell approach to determine what ISGs are early RLR activators and act independently of IFN signaling. They performed smFISH (single molecule fluorescence in situ hybridization) in fixed cells that had been infected with Sendai virus and evaluated host and viral mRNA expression. To prevent ISG activation, Vero cells were used. Due to a genetic defect, Vero cells cannot produce any type of IFN, but have IFNAR and a functional JAK-STAT pathway, so they are still capable of triggering ISGs in response to IFN-B. This work verified the dependence of IRF3/IRF7 on early ISGs (RIG-I, MDA5, LGP2, IFIT1, and OASL) and also calculated the post-infection kinetics of IFN signaling independent gene expression, which occurred as early as three hours post-infection. Furthermore, Doganay et al. correlated ISGs with IFNB1 and each other at the onset of infection. The findings of this study were validated using Nanostring technology using a custom panel of 49 innate immune genes. The validation confirmed that 8 of the 49 innate immune genes were up-regulated (twofold increase) 6-h post-infection without IFNB1 (DDx60, ISG15, IFIT1, LGP2, MDA5, OASL, RIG-I, and Viperin). To validate the dependence of IRF3 on RIG-I and ISG induction, the authors used a siRNA knockout approach on IRF3 and/or IRF7 and qPCR analysis and revealed that RIG-I mRNA was depleted for 9 h post-Sendai virus infection.
The Nanostring platform deployed in Doganay et al. used a digital barcode method that directly hybridizes with user-defined genes with high accuracy and sensitivity. This targeted approach has been employed in other studies to validate novel ISGs in mice (Green et al. 2016; van de Garde et al. 2017) and humans (Chang et al. 2013). Nanostring is a non-amplification-based system that can quantitate expression of roughly 800 genes per sample. This technology competes with traditional qPCR in validating associated ISGs during infection and can function with low yield and degraded RNA.
Computational approaches to deciphering ISG regulation: a transcription factor binding sites approach
In contrast to the above featured studies, alternate studies have focused deciphering the patterns in ISG regulation through transcription binding studies. Understanding the linkage between IRF3 and NFκB is essential to informing us how ISGs relate to disease outcome. Preliminary studies have shown that NFκB production has been tied to TRIF and TNF-α receptor through IRF3 production. Furthermore, NFκB response appears to be dependent on Type I Interferon response (Moschonas et al. 2012) and IFN’s feedback loop through STAT and IRFs (Bertolusso et al. 2014). Iwanaszko et al. used a bioinformatics approach to understand how IRF3 and NF-κB affect each other by identifying overlapping regulatory promoter regions with TFBS (Transcription Factor Binding Sites) (Iwanaszko and Kimmel 2015). Furthermore, the study also looked at the binding site of IFN-ß, identified two ISREs in the promoter region, and found two new co-regulators involved in IRF3 and NF-κB crosstalk: AP-1 (also involved in TLR signaling) and SP1 (involved in NF-κB signaling). Many of the computationally identified binding sites were validated with public ChIPseq data provided through the ENCODE project. During viral infection, the authors determined that NF-κB negatively regulates IRF3, and positively regulates NF-ΚB1, NF-ΚB2, RELA, and REL. They also confirmed regulation in NF-ΚB1, NF-ΚB2, RELA, and AP-1 by NF-κB. This is an elegant example of computational exploratory approach to a problem that could be validated with public data.
Computational approaches to deciphering ISG regulation: an epigenetics approach
In contrast to transcription binding studies, a newer tactic is to study heritable changes surrounding VSG/ISG activation. Downstream of IRF3 and NF-κB activation, researchers are studying the epigenetics of ISG regulation and the mechanisms of chromosomal opening, and the unraveling of gene regulation. Chromatin reorganization has been shown to contribute to the magnitude of IFN induction during virus infection (Huang et al. 2002; Cui et al. 2004; Ni et al. 2005; Yan et al. 2005). As nucleosomes begin remodeling to activate and deactivate genes, they manipulate the chromatin structure, making it either more relaxed or more constricted. This process greatly influences the breadth and overall coverage of VSG or ISG transcription and regulation that occurs during virus infection and response to IFN. Computational approaches linking chromatin modification to gene regulation have been applied to defining VSG and ISG network regulation (Mostafavi et al. 2016; Zhang et al. 1998; Zhu et al. 1999; DaFonseca et al. 2001; Lau et al. 2003).
Machine learning approaches to VSG and ISG discovery
Modern machine learning and deep learning approaches present powerful new platforms to reveal gene regulation where conventional methodologies have limited capacity to define co-regulated VSG or ISG networks. Deep learning (also referred to as neural network analysis) is a computationally intensive form of a machine learning. With the recent advances in cloud computing, and reduction in computer prices, deep learning is now being implemented in bioinformatics and is highly applicable to VSG/ISG studies. Machine learning approaches allow scientists to take advantage of highly complex, multi-dimensional data sets, and visualize patterns and predictions that were impossible with previous tools. Deep learning tools have been utilized in the areas of regulatory genomics (Montgomery et al. 2010; Pickrell et al. 2010), DNA methylation (Gibbs et al. 2010), and epigenetics (Grubert et al. 2015; Waszak et al. 2015), and are fully applicable to describing VSG and ISG regulation. Machine learning also has a strong application in genetic studies whereby an algorithm can be trained to predict loci that influence phenotypic responses to IFN treatment and/or viral infection, thus identifying quantitative trait loci (QTL) that impart gene regulation and define co-expression networks of VSGs and ISGs. QTL studies have been successfully performed in machine learning and deep learning models to identify gene response networks (Kang et al. 2008; Stegle et al. 2010; Parts et al. 2011; Rakitsch and Stegle 2016).
The role of VSGs and ISGs in innate immune programming
Stimulus-induced differentiation of innate and adaptive immune cells leads to a process of immune polarization that defines the direction and breadth of the immune response. Much attention is placed on understanding the dynamics of gene expression that direct immune polarization in specific cell types. For example, transcriptional studies have focused on the segregation of macrophages into functional classifications according to their activation of STAT1 (M1 phenotype) or STAT6 (M2 phenotype), which is important in determining their role in pro-inflammatory (M1) or anti-inflammatory/wound healing (M2) responses directed by macrophages during times of virus infection.
VSGs and ISGs play key roles in the macrophage polarization process (Chistiakov et al. 2018). Early work in macrophage polarization using transcriptional analysis defined a monocyte-macrophage differentiation signature (Becker et al. 2015). This analysis used public datasets to address clinical conditions with two predefined predictive signatures. The signatures were defined by genes induced by IFNγ+LPS, TNFα as a pro-inflammatory, or IL-4 and IL-13 expression signatures. The authors applied these signatures across multiple data sets using different bioinformatics tools (GEO2R, GSEA) using publically available data sets, including gene expression data sets from virus infection and inflammatory responses. Results were then validated with qPCR analyses of gene expression within in vitro experiments. Among those genes identified in the polarization signatures, the first group (IFNγ+LPS, TNFα) contained many known VSGs and ISGs, including OAS, IFITs, IRFs and IFITM1, and IFITM2. Thus, VSGs and ISGs feature roles in innate immune polarization to direct cell effector functions.
In an effort to refine this polarization signature, recent computational studies were performed on gene expression data sets produced from mouse models of macrophage activation. In these studies, researchers performed bioinformatics analysis to examine immune polarization gene signatures in BALB/c murine macrophages undergoing inflammatory responses. Their results identified polarization pathways and gene networks (Jiang et al. 2017) that included VSGs and ISGs, including differentially regulated genes linked with M1 or M2 polarization of macrophages. Suppressors of cytokine signaling (SOC) were among the identified genes. SOCs are ISGs that generate transcriptional programs known to suppress JAK-STAT signaling through the expression of proteins that suppress STAT and JAK activities controlling IFN signaling (Jiang et al. 2017). The study also revealed that SOC genes play a role in governing macrophage phenotype through regulation of ISG expression during the inflammatory response. Similar approaches should be applied to studies of innate and adaptive immune cell subsets to define VSG and ISG signatures that contribute to immune polarization and immune programming.
Computational studies defining ISGs in vaccine response
Understanding how VSGs and ISGs function in immune polarization and programming is paramount for understanding the response to vaccines. Vaccines are the most powerful tool we have for protection against microbial infection, yet the basis of how specific vaccines program the immune response for long-term protection against infection is not understood. Defining the transcriptome and the role of VSG and ISGs in directing the vaccine response is essential for guiding the rationale design of vaccines and vaccine adjuvants. The application of “computational vaccinology” can thus provide valuable insights toward developing well designed vaccines.
Pulendran and colleagues (Querec et al. 2009) applied a systems biology and computational vaccinology approach to define the gene signatures of protection underlying the highly effective yellow fever virus (YFV) vaccine in humans, based on immunization with the YFV YF-17D vaccine. The authors focused on defining innate immune signaling networks and ISG correlates of vaccine protection, wherein they used a variety of bioinformatics tools (DAVID, Transcription factor binding analysis, IPA, and others). To perform a system level analysis, they combined various data types (blood cell gene expression data, cytokines and chemokine profiles, and flow cytometry data sets of lymphocyte subset frequency). Their computational analyses then identified specific genes, including VSGs and ISGs, whose expression linked with a protective vaccine response. To validate these findings, the authors performed a second independent study with different subjects a year later and were able to predict their protective gene signature, using the previous data with 90% accuracy. This was the first major computational vaccinology study, such that subsequent studies now use these approaches as a springboard to define gene and gene network correlates of vaccine protection, thus expanding the role of VSGs and ISGs in immune protection.
A later example of computational vaccinology comes from studies with the Ebola virus outbreak of 2014. Researchers applied bioinformatics approaches to analyze transcriptional data sets generated from studies from nonhuman primates undergoing vaccination with a Vesicular stomatitis virus-based Zaire Ebola virus (EBOV) glycoprotein (VSVΔG/EBOVgp) vaccine (Barrenas et al. 2015). Their objective was to assess the vaccine efficacy and identify VSGs and ISGs that play a role in vaccine protection after virus challenge. Peripheral blood mononuclear cells (PBMCs) were recovered from cynomolgus macaques (CMs) at different time points pre- and post-immunization with VSVΔG/EBOVgp, before and after EBOV challenge. The authors quantified VSGs and ISGs within the host response to vaccination and EBOV infection, using RNA sequencing (RNAseq). The resulting gene expression data sets were compared back to baseline (no vaccine/no EBOV). Animals that were not administered the VSVΔG/EBOVgp succumbed to infection, but those receiving the vaccine were protected against EBOV infection. The investigators performed differential expression as well as functional and network analysis of gene expression on the PBMCs from vaccinated and infected animals. Several ISGs stayed active throughout the time course (DDX58, IFNAR1, IFIT1, IFI27, RSAD2). Their network analysis also revealed the expression of several VSGs and ISGs correlated with vaccine-induced immune activation and immune protection against EBOV. Interestingly, several VSGs and ISGs followed a pattern of vaccine protection, thus linking specific VSGs and ISGs to the VSVΔG/EBOVgp vaccine response.
Other bioinformatics studies have evaluated VSG and ISG expression in vaccination to identify protective gene signatures for sustaining vaccine efficacy as a guide to vaccine design. Forero et al. 2017 evaluated transcriptional differences in human nasal epithelial cultures 24 and 36 h after an exposure to either H3N2 influenza virus (WT) or live attenuated influenza vaccine (LAIV). To identify the protective gene signatures, multiple bioinformatics tools were applied to analyses of the gene expression data set (GSEA, transcriptional factor prediction, microarrays, RNAseq, and correlation analysis). Interestingly, the VSGs and ISGs identified from both groups were influenced by type III IFN induction. Unique to the live attenuated treatment group, the ISG profile linked with enrichment of ISGs in pathways of antigen presentation and effector actions of leukocytes and T lymphocytes. In the LAIV treatment group at 36 h, key VSGs and ISGs (IFI6, IFIT3, ISG15, IFIT1, IFITM1, MX1, IFI35, IRF9, IFITM3, IFITM2, IRF1) were elevated and linked with innate immune protection against infection spread. This study noted the importance of VSGs and type III IFN induction of ISGs in innate immune actions for effective viral control. Together, the studies noted here each reveal that VSGs and ISGs are important contributors to vaccine efficacy and antiviral defense. Approaches to engage specific VSGs and ISGs within rational design of vaccines and vaccine adjuvants hold great promise for the future in generating long-term effective vaccine protection against microbial infection, including emerging viral infection.
Conclusion
Since the discovery of IFN over 60 years ago, IFN actions in infection and immunity are still not fully defined. Identification and validation of VSGs, ISGs, and new classes of ISGs will help advance the immunological field and the design and application of targeted therapeutics. Recent computational and biological advances offer new perspectives that allow us to link transcriptomic profiling, genome analyses, immune programming, and polarization with vaccinology to generate a complete understanding of VSG and ISG function. These resources should include new data driven computational tools for bioinformatics, and the expansion and creation of specialized data set repositories.
References
Barrenas F, Green RR, Thomas MJ, Law GL, Proll SC et al (2015) Next-generation sequencing reveals a controlled immune response to Zaire Ebola virus challenge in cynomolgus macaques immunized with vesicular stomatitis virus expressing Zaire Ebola virus glycoprotein (VSV∆G/EBOVgp). Clin Vaccine Immunol 22(3):354–356
Becker M, De Bastiani MA, Parisi MM, Guma FT, Markoski MM et al (2015) Integrated transcriptomics establish macrophage polarization signatures and have potential applications for clinical health and disease. Sci Rep 5:13351
Bertolusso R, Tian B, Zhao Y, Vergara L, Sabree A et al (2014) Dynamic cross talk model of the epithelial innate immune response to double-stranded RNA stimulation: coordinated dynamics emerging from cell-level noise. PLoS ONE 9(4):e93396
Borden EC, Sen GC, Uze G, Silverman RH, Ransohoff RM et al (2007) Interferons at age 50: past, current and future impact on biomedicine. Nat Rev Drug Discov 6(12):975–990
Chang JJ, Woods M, Lindsay RJ, Doyle EH, Griesbeck M et al (2013) Higher expression of several interferon-stimulated genes in HIV-1-infected females after adjusting for the level of viral replication. J Infect Dis 208(5):830–838
Chistiakov DA, Myasoedova VA, Revin VV, Orekhov AN, Bobryshev YV (2018) The impact of interferon-regulatory factors to macrophage differentiation and polarization into M1 and M2. Immunobiology 223(1):101–111
Chow KT, Gale M Jr (2015) SnapShot: interferon signaling. Cell 163(7):1808–1808.e1
Crow MK (2016) Autoimmunity: interferon α or β: which is the culprit in autoimmune disease? Nat Rev Rheumatol 12(8):439–440
Cui K, Tailor P, Liu H, Chen X, Ozato K et al (2004) The chromatin-remodeling BAF complex mediates cellular antiviral activities by promoter priming. Mol Cell Biol 24(10):4476–4486
DaFonseca CJ, Shu F, Zhang JJ (2001) Identification of two residues in MCM5 critical for the assembly of MCM complexes and Stat1-mediated transcription activation in response to IFN-gamma. Proc Natl Acad Sci USA 98(6):3034–3039
de Veer MJ, Holko M, Frevel M, Walker E, Der S et al (2001) Functional classification of interferon-stimulated genes identified using microarrays. J Leukoc Biol 69(6):912–920
Der SD, Zhou A, Williams BR, Silverman RH (1998) Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc Natl Acad Sci USA 95(26):15623–15628
Dixit E, Boulant S, Zhang Y, Lee AS, Odendall C, Shum B, Hacohen N, Chen ZJ, Whelan SP, Fransen M, Nibert ML, Superti-Furga G, Kagan JC (2010) Peroxisomes are signaling platforms for antiviral innate immunity. Cell 141(4):668–681. https://doi.org/10.1016/j.cell.2010.04.018
Doganay S, Lee MY, Baum A, Peh J, Hwang SY et al (2017) Single-cell analysis of early antiviral gene expression reveals a determinant of stochastic IFNB1 expression. Integr Biol 9(11):857–867
Forero A, Fenstermacher K, Wohlgemuth N, Nishida A, Carter V et al (2017) Evaluation of the innate immune responses to influenza and live-attenuated influenza vaccine infection in primary differentiated human nasal epithelial cells. Vaccine 35(45):6112–6121
Gibbs JR, van der Brug MP, Hernandez DG, Traynor BJ, Nalls MA, Lai SL, Arepalli S, Dillman A, Rafferty IP, Troncoso J, Johnson R, Zielke HR, Ferrucci L, Longo DL, Cookson MR, Singleton AB (2010) Abundant quantitative trait loci exist for DNA methylation and gene expression in human brain. PLoS Genet 6(5):e1000952. https://doi.org/10.1371/journal.pgen.1000952
Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S et al (2002) Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. J Virol 76(11):5532–5539
Green R, Wilkins C, Thomas S, Sekine A, Ireton RC et al (2016) Identifying protective host gene expression signatures within the spleen during West Nile virus infection in the collaborative cross model. Genom Data 10:114–117
Grubert F, Zaugg JB, Kasowski M, Ursu O, Spacek DV, Martin AR, Greenside P, Srivas R, Phanstiel DH, Pekowska A, Heidari N, Euskirchen G, Huber W, Pritchard JK, Bustamante CD, Steinmetz LM, Kundaje A, Snyder M (2015) Genetic control of chromatin states in humans involves local and distal chromosomal interactions. Cell 162(5):1051–1065. https://doi.org/10.1016/j.cell.2015.07.048
Hemann EA, Gale M Jr, Savan R (2017) Interferon lambda genetics and biology in regulation of viral control. Front Immunol 8:1707
Henle W (1950) Interference phenomena between animal viruses: a review. J Immunol 64(3):203–236
Hertzog P, Forster S, Samarajiwa S (2011) Systems biology of interferon responses. J Interferon Cytokine Res 31(1):5–11
Huang M, Qian F, Hu Y, Ang C, Li Z et al (2002) Chromatin-remodelling factor BRG1 selectively activates a subset of interferon-alpha-inducible genes. Nat Cell Biol 4(10):774–781
Hung JH, Yang TH, Hu Z, Weng Z, DeLisi C (2012) Gene set enrichment analysis: performance evaluation and usage guidelines. Brief Bioinform 13(3):281–291
Isaacs A, Lindenmann J, Valentine RC (2015) Pillars article: virus interference II some properties of interferon. Proc R Soc Lond B Biol Sci. 1957. 147:268–273. J Immunol 195(5):1921–1926
Iwanaszko M, Kimmel M (2015) NF-κB and IRF pathways: cross-regulation on target genes promoter level. BMC Genom 16:307
Jiang L, Li X, Zhang Y, Zhang M, Tang Z et al (2017) Microarray and bioinformatics analyses of gene expression profiles in BALB/c murine macrophage polarization. Mol Med Rep 16(5):7382–7390
Kang HM, Ye C, Eskin E (2008) Accurate discovery of expression quantitative trait loci under confounding from spurious and genuine regulatory hotspots. Genetics 180(4):1909–1925. https://doi.org/10.1534/genetics.108.094201
Lachmann A, Torre D, Keenan AB, Jagodnik KM, Lee HJ et al (2018) Massive mining of publicly available RNA-seq data from human and mouse. Nat Commun 9(1):1366
Lau JF, Nusinzon I, Burakov D, Freedman LP, Horvath CM (2003) Role of metazoan mediator proteins in interferon-responsive transcription. Mol Cell Biol 23(2):620–628
Liu L, Wei J, Ruan J (2017) Pathway enrichment analysis with networks. Genes 8(10):246
Montgomery SB, Sammeth M, Gutierrez-Arcelus M, Lach RP, Ingle C, Nisbett J, Guigo R, Dermitzakis ET (2010) Transcriptome genetics using second generation sequencing in a Caucasian population. Nature 464(7289):773–777. https://doi.org/10.1038/nature08903
Moschonas A, Ioannou M, Eliopoulos AG (2012) CD40 stimulates a “feed-forward” NF-κB-driven molecular pathway that regulates IFN-β expression in carcinoma cells. J Immunol 188(11):5521–5527
Mostafavi S, Yoshida H, Moodley D, LeBoité H, Rothamel K et al (2016) Parsing the interferon transcriptional network and its disease associations. Cell 164(3):564–578
Ni Z, Karaskov E, Yu T, Callaghan SM, Der S et al (2005) Apical role for BRG1 in cytokine-induced promoter assembly. Proc Natl Acad Sci USA 102(41):14611–14616
Parts L, Stegle O, Winn J, Durbin R (2011) Joint genetic analysis of gene expression data with inferred cellular phenotypes. PLoS Genet 7(1):e1001276. https://doi.org/10.1371/journal.pgen.1001276
Pickrell JK, Marioni JC, Pai AA, Degner JF, Engelhardt BE, Nkadori E, Veyrieras JB, Stephens M, Gilad Y, Pritchard JK (2010) Understanding mechanisms underlying human gene expression variation with RNA sequencing. Nature 464(7289):768–772. https://doi.org/10.1038/nature08872
Pott J, Stockinger S (2017) Type I and III interferon in the gut: tight balance between host protection and immunopathology. Front Immunol 8:258
Probst P, Grigg JB, Wang M, Muñoz E, Loo YM et al (2017) A small-molecule IRF3 agonist functions as an influenza vaccine adjuvant by modulating the antiviral immune response. Vaccine 35(15):1964–1971
Querec TD, Akondy RS, Lee EK, Cao W, Nakaya HI, Teuwen D, Pirani A, Gernert K, Deng J, Marzolf B, Kennedy K, Wu H, Bennouna S, Oluoch H, Miller J, Vencio RZ, Mulligan M, Aderem A, Ahmed R, Pulendran B (2009) Systems biology approach predicts immunogenicity of the yellow fever vaccine in humans. Nat Immunol 10(1):116–125. https://doi.org/10.1038/ni.1688
Raftery N, Stevenson NJ (2017) Advances in anti-viral immune defence: revealing the importance of the IFN JAK/STAT pathway. Cell Mol Life Sci 74(14):2525–2535
Rakitsch B, Stegle O (2016) Modelling local gene networks increases power to detect trans-acting genetic effects on gene expression. Genome Biol 17:33. https://doi.org/10.1186/s13059-016-0895-2
Rodero MP, Crow YJ (2016) Type I interferon-mediated monogenic autoinflammation: the type I interferonopathies, a conceptual overview. J Exp Med 213(12):2527–2538
Rusinova I, Forster S, Yu S, Kannan A, Masse M et al (2013) Interferome v20: an updated database of annotated interferon-regulated genes. Nucleic Acids Res 41(Database issue):D1040–D1046
Samarajiwa SA, Forster S, Auchettl K, Hertzog PJ (2009) INTERFEROME: the database of interferon regulated genes. Nucleic Acids Res 37(Database issue):D852–D857
Sarkar SN, Sen GC (2004) Novel functions of proteins encoded by viral stress-inducible genes. Pharmacol Ther 103(3):245–259
Schneider WM, Chevillotte MD, Rice CM (2014) Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32:513–545
Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT et al (2011) A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472(7344):481–485
Schoggins JW, MacDuff DA, Imanaka N, Gainey MD, Shrestha B et al (2014) Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 505(7485):691–695
Schroder K, Hertzog PJ, Ravasi T, Hume DA (2004) Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol 75(2):163–189
Shaw AE, Hughes J, Gu Q, Behdenna A, Singer JB et al (2017) Fundamental properties of the mammalian innate immune system revealed by multispecies comparison of type I interferon responses. PLoS Biol 15(12):e2004086
Sohn JI, Nam JW (2018) The present and future of de novo whole-genome assembly. Brief Bioinform 19(1):23–40
Stark GR, Darnell JE Jr (2012) The JAK-STAT pathway at twenty. Immunity 36(4):503–514
Stegle O, Parts L, Durbin R, Winn J (2010) A Bayesian framework to account for complex non-genetic factors in gene expression levels greatly increases power in eQTL studies. PLoS Comput Biol 6(5):e1000770. https://doi.org/10.1371/journal.pcbi.1000770
van de Garde MDB, Pas SD, van Oord GW, Gama L, Choi Y et al (2017) Interferon-alpha treatment rapidly clears Hepatitis E virus infection in humanized mice. Sci Rep 7(1):8267
Waszak SM, Delaneau O, Gschwind AR, Kilpinen H, Raghav SK, Witwicki RM, Orioli A, Wiederkehr M, Panousis NI, Yurovsky A, Romano-Palumbo L, Planchon A, Bielser D, Padioleau I, Udin G, Thurnheer S, Hacker D, Hernandez N, Reymond A, Deplancke B, Dermitzakis ET (2015) Population variation and genetic control of modular chromatin architecture in humans. Cell 162(5):1039–1050. https://doi.org/10.1016/j.cell.2015.08.001
Webster B, Assil S, Dreux M (2016) Cell-cell sensing of viral infection by plasmacytoid dendritic cells. J Virol 90(22):10050–10053
Yan Z, Cui K, Murray DM, Ling C, Xue Y et al (2005) PBAF chromatin-remodeling complex requires a novel specificity subunit, BAF200, to regulate expression of selective interferon-responsive genes. Genes Dev 19(14):1662–1667
Zhang JJ, Zhao Y, Chait BT, Lathem WW, Ritzi M et al (1998) Ser727-dependent recruitment of MCM5 by Stat1alpha in IFN-gamma-induced transcriptional activation. EMBO J 17(23):6963–6971
Zhu M, John S, Berg M, Leonard WJ (1999) Functional association of Nmi with Stat5 and Stat1 in IL-2- and IFNgamma-mediated signaling. Cell 96(1):121–130
Acknowledgements
Supported by National Institutes of Health Grants U19AI100625, R01AI104002, and U19AI083019. The funders had no role in the design, data acquisition, analysis, or preparation of the manuscript. Thanks to Amy Green for proof reading. Thanks to Adrianna Forero for her support and critical insights.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
Rights and permissions
About this article

Cite this article
Green, R., Ireton, R.C. & Gale, M. Interferon-stimulated genes: new platforms and computational approaches. Mamm Genome 29, 593–602 (2018). https://doi.org/10.1007/s00335-018-9755-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-018-9755-6