
Abstract
Considerable clinical and molecular variations have been known in retinal blinding diseases in man and also in dogs. Different forms of retinal diseases occur in specific breed(s) caused by mutations segregating within each isolated breeding population. While molecular studies to find genes and mutations underlying retinal diseases in dogs have benefited largely from the phenotypic and genetic uniformity within a breed, within- and across-breed variations have often played a key role in elucidating the molecular basis. The increasing knowledge of phenotypic, allelic, and genetic heterogeneities in canine retinal degeneration has shown that the overall picture is rather more complicated than initially thought. Over the past 20 years, various approaches have been developed and tested to search for genes and mutations underlying genetic traits in dogs, depending on the availability of genetic tools and sample resources. Candidate gene, linkage analysis, and genome-wide association studies have so far identified 24 mutations in 18 genes underlying retinal diseases in at least 58 dog breeds. Many of these genes have been associated with retinal diseases in humans, thus providing opportunities to study the role in pathogenesis and in normal vision. Application in therapeutic interventions such as gene therapy has proven successful initially in a naturally occurring dog model followed by trials in human patients. Other genes whose human homologs have not been associated with retinal diseases are potential candidates to explain equivalent human diseases and contribute to the understanding of their function in vision.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The past 20 years have seen a virtual explosion in gene discovery of inherited retinal diseases (RDs) in humans (Fig. 1). In man, these are clinically heterogeneous, characterized by defects in different retinal cell types, variable in age of onset, and diverse degrees of severity. These inherited diseases, subdivided by aspects of the clinical phenotype observed, include retinitis pigmentosa (RP), cone–rod dystrophy (CRD), and Leber congenital amaurosis (LCA), among others.

Accumulation of the numbers of mapped loci (light gray) and identified genes (dark gray) in human RDs from 1980 to 2011. Reprinted from RetNet, http://www.sph.uth.tmc.edu/RetNet, with permission (Stephen P. Daiger, PhD, and the University of Texas Health Science Center at Houston)
Since the mapping of an autosomal dominant RP locus to the RHO (rhodopsin) gene (Farrar et al. 1990; McWilliam et al. 1989), at least 213 retinal disease loci/172 genes have been independently associated with human RDs (RetNet, http://www.sph.uth.tmc.edu/RetNet/) (Supplementary Fig. 1). In some of these genes, over 100 mutations have been identified. Such extensive genetic and allelic heterogeneities likely reflect the observed clinical heterogeneity.
Dogs also suffer from various forms of naturally occurring RDs causing blindness, with the most common known disease class in the veterinary field being progressive retinal atrophy (PRA). Over 100 breeds are affected with PRA, and all affected dogs show clinically recognizable abnormalities and behavioral signs indicative of visual deficits at various ages and with different degrees of progression. Since PRA was identified as a clinical entity by Magnusson in the early 20th century, it was recognized as the homolog to RP in humans, and the diseases in both species shared genetic, clinical, and pathological features (Magnusson 1909, 1910, 1911, 1917). Remarkable early progress in establishing the dog as a model for RD research was accomplished by Parry in a series of seminal studies published in the mid-20th century (Parry 1953a, b, 1954a, b; Parry et al. 1953, 1955), and the history and the evolution of research in this field has been summarized more recently (Aguirre and Acland 2006).
Pedigree dog breeds have unique breeding histories over the past 100–300 years, with strong effects from popular sires as well as breed barriers. As a result, most dogs of the same breed are relatively uniform genetically, just like certain isolated human populations in which many sequence variants are shared across the population. This within-breed genetic uniformity has been advantageous in studies of canine genetic traits, including RDs. While human RDs encompass a broad phenotypic and genetic spectrum making it difficult to isolate and dissect each form, the diseases within a dog breed often are phenotypically and genetically uniform and thus more tractable. With canine RDs considered homologs to those of man, molecular studies in dogs have continued to contribute to the understanding of the human diseases.
The within-breed phenotypic and genetic uniformities have contributed much to genetic studies of canine RDs and, at the same time, served to highlight heterogenic characteristics suggestive of additional diseases in a breed. For example, genetic heterogeneity is seen in Golden retrievers which were thought to be affected with progressive rod–cone degeneration (prcd) only, but at least two other forms of PRA, including GR_PRA1, are recognized now in the breed (Downs et al. 2011). However, the phenotypic and genetic heterogeneities within the same RD form, as well as within a breed, are not as extensive or complex as in human populations, making it possible to carry out gene discovery studies once the phenotypes and genetics are properly ascertained.
In this article we review the molecular participants involved in the complex retinal disease system, classify the clinically distinct forms of canine RDs, and trace the different approaches developed and applied for disease gene discovery. We dissect the heterogenic nature that has become increasingly evident in canine RDs, showing examples of its effective application to gene discovery.
Structural and functional elements of the retina: a key to understanding diseases and pathways
The retina
Dogs are born with an undeveloped retina that reaches maturity at 3–6 weeks postnatally (Aguirre et al. 1972; Parry 1953a), whereas humans are born with a retina that is much further developed and undergoes further postnatal differentiation, particularly of the fovea and macula (Abramov et al. 1982). Nevertheless, the structure of the mature canine retina is broadly similar to that of man, consisting of layers of specialized cells (Fig. 2a, b). The outermost layer, comprising the light-sensitive rod and cone photoreceptors, abuts and is nourished by the retinal pigment epithelium (RPE). Each photoreceptor has outer (OS) and inner (IS) segments. Phototransduction is initiated on the stacked disks of the OS while the biosynthetic functions are concentrated in the IS. The connecting cilium, bridging the OS and the IS, has an important role in intraflagellar transport (see Webvision for further details: http://webvision.med.utah.edu/). Of the photoreceptor cells, cones are responsible for day and color vision, whereas rods confer night or dim light vision. In both humans and dogs, rods and cones are unevenly distributed across the retina and the number of rods by far exceeds the number of cones (Curcio et al. 1990; Mowat et al. 2008).

The structural and functional elements of the retina and the localization of selected genes involved in canine retinal degeneration. a Histologic section of the canine retina showing the highly ordered lamination from the outer (top) to the inner (bottom) retina. The rod and cone photoreceptors that form the outermost layer receive the light stimuli and are nourished by the retinal pigment epithelium (RPE). The outer nuclear layer (ONL) contains the nuclei and cell bodies of the photoreceptors. The inner nuclear layer (INL) contains the nuclei of the secondary neurons (i.e., bipolar, amacrine, and horizontal cells) and the glial Müller cells. The innermost ganglion cell layer (GCL) receives the input from photoreceptors via the bipolar and amacrine cells and transmit the signals to the brain. b Localization of selected genes associated with RDs in dogs. *Indicates the cone photoreceptors. Canine RD genes with unknown retinal localization or ubiquitous expression are not displayed. OS outer segment, IS inner segment. c The retinoid cycle recycles the light-absorbing chromophore. Absorption of a photon (hν) converts 11-cis-retinal bound to opsin (Rho) into all-trans-retinal initiating phototransduction. All-trans-retinal is reduced by photoreceptor retinol dehydrogenase (RDH) to all-trans-retinol and exported to the RPE where it is esterified by lecithin retinol acyltransferase (LRAT), converted to 11-cis-retinol by RPE65, and oxidized to 11-cis-retinal by NAD and a cis-specific retinol dehydrogenase (cis-RDH). 11-cis-retinal is exported back to the OS to again bind to opsin. CRALBP cellular retinaldehyde-binding protein, CRBP cellular retinol-binding protein, IRBP interphotoreceptor matrix retinoid-binding protein. d The phototransduction cascade converts light stimuli to electrical signal. Activated opsin (R*) in the disk membrane activates transducin (G) to G* which in turn activates phosphodiesterase (PDE) to PDE**. PDE** hydrolyzes cGMP, reducing its cytoplasmic concentration which results in closure of cGMP-gated channels in the plasma membrane. This causes hyperpolarization of the photoreceptors leading to signals sent to the downstream neurons. The schematics are modified and reprinted with permission from John Wiley and Sons (Nawrot et al. 2006) (c) and Elsevier Limited (Leskov et al. 2000; Pugh 1999) (d)
Two critical pathways involved in vision are the retinoid cycle (Fig. 2c) and phototransduction (Fig. 2d). The phototransduction cascade is triggered by the absorption of light photons and generates an amplified chemical signal. All photoreceptor OSs contain an opsin bound to a light-absorbing chromophore, 11-cis-retinal, the precursor of which is all-trans-retinol (vitamin A). The opsins in rods (rhodopsin) and cones (photopsin) differ significantly in their spectral sensitivity, i.e., the range of wavelengths to which they are most sensitive. The retinoid cycle recycles all-trans-retinol to 11-cis-retinal in the RPE and replenishes the chromophore for rod and cone opsin pigments. Mutations in genes participating in the activation or deactivation of phototransduction as well as the retinoid cycle are causally associated with RDs. Many of the known genes underlying canine retinal diseases code for proteins localized to the outer retina where these functions take place (Fig. 2b).
Genes associated with RDs in man and dogs
Although mutations in phototransduction or visual cycle genes have repeatedly been identified as causes of RDs, they represent but a small part of the suite of disease-associated genes that cause blindness. Human RDs are known for extensive phenotypic and genetic heterogeneities (Supplementary Table 1) resulting in disease categories such as RP, CRD, and LCA. RP is associated with initial night blindness and subsequent progressive loss of peripheral visual fields. In contrast, the cones and central vision are initially more affected in CRD, while LCA manifests in childhood, with severe visual impairment or total blindness. Syndromic forms of RD (e.g., sensorineural blindness and deafness represented by Usher syndrome disease class) involve mutations in genes common to ciliated sensory hair cells and photoreceptors (van Wijk et al. 2006, 2009). While canine RDs have been identified in the RP, CRD, and LCA disease classes, syndromic RDs have not been identified (Supplementary Table 1). It is possible that lack of recognition could result in the different expression of the auditory/vestibular and retinal phenotypes with mutations in Usher syndrome genes, as occurs in mice where mutations in almost all of the Usher syndrome genes cause vestibular and auditory abnormalities but minimal or no retinal phenotypes.
The clinical manifestations and the underlying genes vary substantially within each disease category. On the other hand, the same gene may be associated with multiple forms of RD showing genotypic as well as phenotypic overlap among them. For example, recessive mutations in GUCY2D cause LCA, whereas dominant mutations in the same gene result in CRD (Booij et al. 2005; Hunt et al. 2010). In accordance with the homology in the retinal system, almost all known canine genes underlying different forms of RD have human disease counterparts (outlined or underlined in Supplementary Table 1). Also, all the genes associated with human RDs are potential candidates for the equivalent canine condition.
Inherited retinal diseases in dogs: diverse phenotypic groups
Retinal diseases in dogs can be classified as progressive, stationary, or developmental (Table 1). While the majority of canine RDs are autosomal recessive, other forms of Mendelian inheritance are known: an autosomal dominant PRA (ADPRA) is known to be in the Bullmastiff and English mastiff dogs with a missense mutation in (RHO) gene (Kijas et al. 2002, 2003). X-linked forms of PRA in Samoyeds and Siberian Huskies (XLPRA1) and in mixed breeds (XLPRA2) (Acland et al. 1994; Zhang et al. 2002) are known to be caused by nonallelic mutations in the RPGR (retinitis pigmentosa GTPase regulator) gene.
Progressive disorders
Progressive forms of canine RDs affect primarily the photoreceptor cells and are classified as PRA and CRD. The progressive designation is based on serial clinical and functional assessments that show a continuous loss of retinal structure over time accompanied by loss of electroretinogram (ERG) rod and/or cone function. The rate of progression is variable and gene/mutation- and breed-dependent.
PRA, homologous to RP in humans, is characterized by initial degeneration of rod photoreceptors. The mutations can affect rods only (e.g., PDE6B in rcd1), or both rods and cones (e.g., RPGR in XLPRA1 and 2), but earlier and more severe loss of rods is the salient PRA phenotype (Fig. 3a). Consequently, cone loss can be secondary and the result of the rod degeneration, or it can be a primary event that occurs later and progresses more slowly than the rod disease. The dominance of rod involvement in PRA causes night blindness, commonly the initial clinical finding that progresses to severe visual dysfunction under both dim and bright light. Associated with these defects, affected dogs show characteristic fundus changes that are visible with an ophthalmoscope—hyperreflectivity of the tapetum, attenuation of the retinal blood vessels, and pale optic discs. Some dogs may develop secondary cataracts in the late stages of the disease.

ERG of dogs affected with PRA and CRD. Rod, rod/cone, and cone ERG recordings obtained by dark-adapted responses to blue, red, and white flashes, respectively. Rod and cone flicker recordings were obtained by low- (5 Hz) and high-intensity [5 Hz (a), 30 Hz (b)] white flashes, respectively. Onset of stimuli for the rod and cone flicker responses are indicated by the upward deflection of square wave pulses below the responses (a) or short vertical arrows under the responses (b). a Absence of rod responses with subsequent and progressive deterioration of the cone function in a Norwegian elkhound affected with rd (reprinted from Aguirre 1978). b Cone dysfunction detected by 1.2 years, followed by further deterioration of both cone and rod function in Glen of Imaal terrier dogs affected with crd3. At all ages, the loss of cone function is more prominent than that for rods (reprinted with permission from Goldstein et al. 2010c)
In contrast, CRD is a disorder predominantly of cones, with rods being affected later or to a lesser extent, at least initially. Particularly in early-onset forms, this can cause the initial clinical signs to be defects in photopic (day) visual function. Many cases, however, are referred when there is already extensive impairment of visual function under both bright and dim light, and the ophthalmoscopic findings are similar to PRA. Hence, the identification of a cone–led abnormality relies on ERG testing at the early stage of disease pathology (Kijas et al. 2004; Turney et al. 2007) (Fig. 3b). For this reason, CRD may be indistinguishable from PRA at the clinical level, thus often being referred to as PRA, although it is a distinct disorder by itself. Most of the currently recognized CRD forms, e.g., crd1 in American Staffordshire terriers, crd2 in American pit bull terriers, cord1 (crd4) in Miniature longhaired dachshunds, and CRD in Standard wirehaired dachshunds, show extensive retinal degeneration before 1 year of age (Curtis and Barnett 1993; Kijas et al. 2004; Ropstad et al. 2007, 2008), while crd3 in Glen of Imaal terriers manifests later in life with slower progression (Goldstein et al. 2010c; Kropatsch et al. 2010).
Clinical studies of PRA/CRD have shown a broad age-of-onset range in various breeds. Although affected dogs show similar ophthalmoscopic abnormalities at the end stage of the disease, the age when the first changes become evident and the rate of progression vary, and is mostly breed-specific (Supplementary Table 2). Retinal dysfunction detected by reduction in ERG amplitudes has long been the only available tool to detect subclinical PRA/CRD, as ophthalmoscopic abnormalities may not be detectable until at least 70% of the photoreceptor cells have been lost (Parshall et al. 1991).
Early-onset PRA/CRD manifests during the postnatal retinal differentiation period (2–6 weeks) and results from abnormal or arrested retinal development or progressive degeneration that commences during or immediately after retinogenesis. The early onset of these diseases typically results in relatively rapid progression toward end-stage retinal degeneration and can be clinically evident in young adult or preadult dogs (Supplementary Table 2).
In contrast, late-onset PRA/CRD is characterized by pathological changes that first become evident after normal development and maturation of the retina. As such, they represent defects in pathways critical for the long-term maintenance of normal photoreceptor function and viability. Visual–behavioral abnormalities may not become apparent until well after reproductive maturity. Typically, further progression toward end-stage retinal degeneration is much slower than in early-onset PRA/CRD and may not become clinically evident until very late in life.
Stationary disorders
In contrast to the progressive ophthalmoscopic changes and visual–behavioral problems observed in PRA/CRD, certain forms of RD show either extremely slow or no further abnormalities or deterioration in vision from the time of initial diagnosis in young animals. Both canine achromatopsia, previously known as cone degeneration and canine hemeralopia (Aguirre and Rubin 1974, 1975), in the Alaskan Malamute (cdAMAL) and the German shorthaired pointer (cdGSPT) (Sidjanin et al. 2003), and canine LCA (cLCA), previously known as congenital stationary night blindness (csnb) (Narfström et al. 1989) and retinal dystrophy (Wrigstad 1994) in the Briard, are essentially stationary disorders. Cones alone are affected in cdAMAL and cdGSPT because of a mutation in CNGB3, a cone–specific gene, whereas both rods and cones are dysfunctional in cLCA because a mutation in RPE65, a RPE-specific isomerohydrolase, impairs the retinoid cycle and 11-cis-retinal is not made (Fig. 2c). While the achromatopsia-affected retina shows no further deterioration with age in vision or retinal abnormalities, old cLCA-affected dogs show retinal thinning, both clinically and histologically, an indication that the stationary classification may be a misnomer.
Developmental disorders
Developmental disorders affect retinal development and are considered nondegenerative retinal diseases. Retinal dysplasia is a developmental disorder characterized by a defect in retinal differentiation. Nonsyndromic forms have been reported in several breeds [e.g., Labrador retrievers (Barnett et al. 1970) and Bedlington terriers (Rubin 1968)], but the underlying genetic causes are unknown. On the other hand, syndromic retinal dysplasia in Labrador retrievers (osd1) and Samoyeds (osd2) are nonallelic oculoskeletal disorders that model autosomal recessive Stickler syndrome in man; they are caused by mutations in COL9A3 and COL9A2, respectively, both of which code for components of the type IX collagen fibril (Goldstein et al. 2010a; Meyers et al. 1983; Nelson and MacMillan 1983).
Another developmental disorder is Collie eye anomaly (cea) which affects the retina–choroid–scleral complex and serves as a model for macular coloboma in man. Disease phenotype is variable, ranging from choroidal hypoplasia, scleral coloboma, and retinal detachment. Because of the high disease prevalence of cea in Collies on a worldwide basis (70–90%), it is the most common inherited canine retinal disorder, although most affected dogs are not visually impaired (Barnett and Stades 1979; Roberts 1969). Shared ancestry among Collie breeds, including the Rough and Smooth collie, Australian shepherd, Border collie, and Shetland sheepdog, allowed the fine mapping of a region in the canine chromosome 37 (CFA37) to a common disease haplotype, and identified a homozygous deletion in NHEJ1 that spans a highly conserved binding domain (Lowe et al. 2003; Parker et al. 2007).
Searching for genes and mutations that cause canine retinal degenerations
The approaches used to search for genes and mutations underlying inherited traits of the dog have evolved with the availability of canine genomic resources which initially were negligible; for the most part, no canine-specific resources were available prior to 1997. Initial attempts focused on candidate genes, with the shift to linkage mapping and positional cloning strategies (Fig. 4). More recently, the microarray-based genome-wide association study (GWAS) with SNPs has been used as it became available.

Discovery approaches for canine RD loci and genes. The cumulative numbers of loci and mutations identified as causing canine RD are shown, classified according to the discovery approach used. Note that the chromosomal location and the mutation were reported concurrently for cd, cord1, osd1, and osd2. The disease symbols correspond to those shown in Table 1
The initial tools for mapping were provided by the canine meiotic linkage map (Mellersh et al. 1997), dog–rodent somatic hybrid cell lines (Langston et al. 1997), low- and high-resolution radiation hybrid maps (Guyon et al. 2003; Priat et al. 1998) that were integrated with single-locus probes (Breen et al. 2001), and BAC library (Li et al. 1999). Following the publication of a 1.5X genome sequence of the poodle (Kirkness et al. 2003), the 7.5X sequence of the Boxer dog became available in 2005 along with a dense map of single nucleotide polymorphisms (SNPs) across breeds (Lindblad-Toh et al. 2005).
Among the different canine diseases, RDs may have had the most comprehensive variety of approaches used to identify the underlying genes and mutations. The approaches developed and tested are described below in chronological order.
Candidate gene analysis
Early studies relied on a candidate gene approach largely because of the lack of alternative canine genomic tools. Candidate genes were most commonly selected for evaluation based on a priori knowledge of the biochemical pathway or defect involved, or on information from other species such as humans and mice. Novel candidates were also identified by the use of subtraction techniques to clone genes differentially expressed between normal and mutant retinas (Zangerl et al. 2002; Zhang et al. 1998).
The first mutation responsible for PRA was found by screening PDE6B as a candidate gene for rcd1 in Irish setters (Clements et al. 1993; Suber et al. 1993), because this canine disease was phenotypically and biochemically similar to rd in mice resulting from a PDE6B mutation (Bowes et al. 1990; Farber and Lolley 1974). In rcd1, the activity of retinal cyclic guanosine monophosphate phosphodiesterase (cGMP-PDE) was impaired (Aguirre et al. 1978, 1982b), and the mRNA levels for PDE6B were selectively decreased as in rd mice (Farber et al. 1992). Sequencing of canine PDE6B identified a stop mutation in exon 21 (Clements et al. 1993; Ray et al. 1994; Suber et al. 1993). A candidate gene approach was also used to successfully identify a different mutation in PDE6B for rcd1a in the Sloughi (Dekomien et al. 2000), PDE6A for rcd3 in the Cardigan Welsh corgi (Petersen-Jones et al. 1999), and RHO for ADPRA in the Bullmastiff and English mastiff (Kijas et al. 2002).
However, many canine RDs were tested for mutations in many different candidate genes related to retinal structure or function without success. Examples include PDE6A (Wang et al. 1999), PDE6D and PDE6G (Dekomien and Epplen 2003; Wang et al. 1999), arrestin (Dekomien and Epplen 2002c), peripherin/rds (Ray et al. 1996; Runte et al. 2000), ROM1 (Gould et al. 1997; Klein et al. 1998), phosducin (Dekomien and Epplen 2002a; Lin et al. 1998; Zhang et al. 1998), opsin (Gould et al. 1995; Ray et al. 1999), and recoverin (Dekomien and Epplen 2002b). As mutations could be identified in only 3.4% of the candidate gene studies of canine RDs, there was considerable pessimism as to the applicability and efficacy of this approach (Aguirre-Hernandez and Sargan 2005). Given that many negative results for these studies may have never been published, the success rate could be even lower.
Nonetheless, the “classic” candidate gene approach has recently proven successful in identifying the mutations in BEST1 associated with cmr1, cmr2, and cmr3 where a phenotype-directed candidate gene approach was used (Guziewicz et al. 2007; Zangerl et al. 2010). Canine multifocal retinopathy (cmr) shows initial multifocal areas of retinal elevation that closely resemble those of the vitelliform or yolk-like lesion in Best macular dystrophy (BMD) human patients. Both canine and human forms eventually progress into retinal atrophy with concomitant histopathologic RPE abnormalities. Screening of BEST1, known to be associated with BMD, promptly identified mutations in cmr dogs, underscoring the clinical, pathologic, and molecular resemblances between the homologous canine and human diseases.
Linkage mapping and positional cloning strategies
Unlike candidate gene studies, which limit the genomic region of inspection, a whole-genome linkage analysis (LA) mapping interrogates the entire genome, including unknown genes, using informative families containing affected and unaffected dogs. Cosegregation of genetic markers and phenotype is analyzed to identify recombination, or lack thereof, between the disease phenotype and marker alleles. Subsequently, the region of interest is fine-mapped using additional markers to increase the resolution, and positional candidate genes in the narrowed interval are analyzed.
To date, 11 chromosomal loci have been mapped by LA leading to the identification of 12 mutations corresponding to different forms of RD. In many cases, identification of the actual gene/mutation took some time after mapping of the chromosomal location. This was due to the long linkage disequilibrium (LD) that had facilitated the mapping process but then ironically hindered narrowing of the critical interval and the number of genes within. One example is the prcd-PRA locus mapped on CFA9 using multiple informative pedigrees (Acland et al. 1998). Genetic markers linked to the disease-causing mutation in the mapped interval were used to perform a “linked” DNA test until the corresponding mutation was identified by positional cloning of a novel gene PRCD (Zangerl et al. 2006).
In LA mapping, large and complete multigenerational canine pedigrees with phenotypic data are required. In theory, the dog is suitable for LA mapping in that detailed pedigree records, large-sized families, and condensed life span facilitate sample collection from multiple generations. In practice, however, accessing the sample population to ascertain phenotypes and eliminate this variable, together with access to all necessary DNA samples, is often a daunting task. For that reason, many of the mapping studies have relied on purpose-bred research colonies developed from naturally occurring cases and expanded by backcrossing and/or outcrossing. Not only can the phenotype be ascertained using multiple testing modalities, but samples from the target and other tissues are readily obtained for analysis to validate gene expression, positional candidates, and phenotype–genotype correlation (for review see Aguirre and Acland 2006).
Genome-wide association studies
As part of the project that developed the 7.5X canine genome reference sequence, over 2.5 million SNPs were identified across the canine genome (Lindblad-Toh et al. 2005). These markers are more abundant than microsatellites and sufficiently densely and broadly placed across the genome to enable association studies. Furthermore, genotyping microarrays have become available, making high-throughput SNP analysis feasible in both humans and domestic animals (Andersson 2009). For the dog, SNP platforms that yield 22,362 (22 k) (Illumina), 49,663 (50 k) (Affymetrix), 127 k (Affymetrix), and 170 k (Illumina) SNP genotypes are available.
A decided advantage of case-control GWAS over LA is that the former does not necessarily require the large, multigenerational pedigrees which, in most cases, require purpose-bred research colonies. Based on the long within-breed LD (0.5–5 Mb, depending on the breed) (Gray et al. 2009; Sutter et al. 2004), 10,000–30,000 SNPs are considered sufficient to map a Mendelian trait with high penetrance and no phenocopies. With this marker density, 20 cases and 20 controls and 50 cases and 50 controls are considered more than sufficient to map autosomal recessive and dominant traits, respectively (Lindblad-Toh et al. 2005).
Since the early successful GWAS, which identified the hair ridge locus in Rhodesian ridgebacks (Karlsson et al. 2007; Salmon Hillbertz et al. 2007) and the white spotting locus in Boxers and other breeds (Karlsson et al. 2007), over 25 genetic traits ranging from ocular (Kuchtey et al. 2011), neurologic (Awano et al. 2009; Drogemuller et al. 2010; Farias et al. 2011; Zeng et al. 2011), dermatologic/immunologic (Drogemuller et al. 2008; Olsson et al. 2011; Wang et al. 2011; Wilbe et al. 2010; Wood et al. 2009), orthopedic (Drogemuller et al. 2009; Parker et al. 2009; Zhou et al. 2010), cardiologic (Meurs et al. 2010), and behavioral disorders (Dodman et al. 2010) to morphology (Bannasch et al. 2010; Boyko et al. 2010) and coat types (Cadieu et al. 2009) have been mapped by GWAS. For canine RDs, three disease forms have been mapped by GWAS to date validating this approach.
Autosomal recessive CRD in the Standard wirehaired dachshund was mapped using an array-based genome-wide sibling transmission disequilibrium test (sibTDT) approach with 13 discordant sib-pairs from a purpose-bred research colony (Wiik et al. 2008). Using 50 k SNP chips, the CRDSWHD locus was mapped to a 6.5-Mb interval on CFA5 containing <70 genes. Fine mapping using 36 informative offspring could not narrow the critical interval. Resequencing of positional candidate genes identified a 180-bp deletion in exon/intron 5 of NPHP4 (nephronophthisis 4). RNA analysis of NPHP4 showed that skipping exon 5 resulted in a truncated protein lacking the domain interacting with RPGRIP1 in the retina but retention of the domain interacting with NPHP1 in the kidney. As mutations in NPHP4 had been associated with simultaneous eye and kidney disease in humans and mice (Mollet et al. 2002; Otto et al. 2002), the lack of the RPGRIP1-interacting domain and retention of the NPHP1-interacting domain explain the retina-specific clinical signs in CRDSWHD.
A second example is the autosomal recessive crd3 in the Glen of Imaal terrier (Goldstein et al. 2010c). Mostly client-owned and chosen to be the least related, 20 affected cases and 22 unaffected controls were selected for a case-control GWAS using 127 k SNP chips. The crd3 locus was mapped to a 2.74-Mb region of homozygosity on CFA16. Sequencing of positional candidate genes identified a large genomic deletion of approximately 23 kb, including exons 15 and 16 of ADAM9. The identification of an ADAM9 mutation established crd3 as the canine ortholog of CORD9, a form of human CRD (Parry et al. 2009).
The most recent demonstration of GWAS in a canine RD was GR_PRA1 in the Golden retriever (GR) (Downs et al. 2011). RD caused by the canine prcd mutation is known to segregate in GRs, but identification of PRA-affected cases lacking the prcd mutation prompted an association study. GWAS using 22 k SNPs on 27 cases (RD-affected, non-prcd) and 19 controls from the GR pet population mapped the GR_PRA1 locus to a 644-kb interval of homozygosity on CFA37. Sequencing positional candidate genes identified a 1-bp insertion in SLC4A3 (solute carrier family 4, anion exchanger, member 3) that resulted in a premature stop codon. Although the mutation cosegregated with the PRA phenotype in certain lineages with autosomal recessive mode of inheritance, there was substantial genotype–phenotype discordance in the overall GR population studied suggesting the presence of one or more additional PRA loci. Although SLC4A3 seems to have a role in retinal function, it has not previously been associated with RD in human patients or naturally occurring mouse strains, making GR_PRA1 a unique model for further investigation.
Effective use of isolated subpopulations
prcd in different breed populations
The current approach of choice to identify the genes and the mutations underlying canine RDs is a SNP chip-based GWAS followed by fine mapping and sequencing of positional candidates. However, narrowing of the mapped critical region to a manageable interval remains challenging. While the extensive within-breed LD facilitates initial mapping of a chromosomal region, moving to the target gene may be hindered by the same phenomenon, with LD extending for megabases and spanning over 100 genes. The prcd locus was initially mapped by LA to a 1.9-Mb interval on CFA9 and contained about 50 genes. Identification of rare recombinant dogs and generation of a unique disease-specific haplotype using SNPs from BAC clones reduced the disease interval. Moreover, as the disease was shown to be present in many different breeds, ancestral recombinations were identified in different breeds (i.e., genetically isolated populations) to establish a narrower disease-specific LD region. Fine mapping eventually narrowed the LD interval to 87 kb containing three genes (Goldstein et al. 2006) leading to the identification of a point mutation in a novel gene, PRCD (Zangerl et al. 2006). This effort was complemented by a parallel approach, exploiting retinal expressed sequence tag (EST) characterization that mapped an EST clone to the disease interval (Zangerl et al. 2009) that proved to be part of the PRCD transcript.
cord1 in miniature longhaired dachshunds: differences in research and pet populations
Even if a particular form of RD is apparently restricted to a single breed, artificially and/or geographically isolated subpopulations of that breed can be exploited to aid in genetic analyses. The autosomal recessive, early-onset (<1 year) form of RD in Miniature longhaired dachshunds (MLHDs) was characterized using a research colony developed in the UK (Curtis and Barnett 1993). The disease was subsequently shown to be CRD and termed cord1 (cone–rod dystrophy 1) (Mellersh et al. 2006; Turney et al. 2007).
Affected cases and obligate carriers from the research colony were used to map the cord1 locus, and a whole-genome scan based on homozygosity and subsequent LA identified a 14.15-Mb interval on CFA15 (Mellersh et al. 2006). The human syntenic region contained a strong candidate gene, RPGRIP1 (RPGR-interacting protein 1), which had been associated with LCA and CRD in humans (Dryja et al. 2001; Gerber et al. 2001; Hameed et al. 2003). Sequencing of RPGRIP1 revealed a homozygous 44-bp insertion (RPGRIP1 −/−) in affected dogs resulting in a frameshift, and a premature stop codon that truncated most of the protein’s functional domain. The mutation segregated completely with cord1 in the research colony.
Meanwhile, Miniature dachshunds had enjoyed an explosive popularity in Japan, with over one million new registrations between 1999 and 2008 according to the Japan Kennel Club. The sudden growth of the breed’s population from a relatively small number of founders led to frequent manifestation of recessive diseases, including RDs. When samples from MLHD with RD collected in Japan were examined, it was found that the age of onset ranged broadly from 4 months to 15 years (mean ± SD = 4.6 ± 3.4 years), a striking difference from the UK research dogs that showed a uniform age of onset (Miyadera et al. 2009). There was also significant discordance between the retinal phenotype and the RPGRIP1 genotype in the broader MLHD population: 20% of blind cases were not RPGRIP1 −/−, while 16% of the apparently normal dogs were, in fact, homozygous mutant for the 44-bp insertion. Nonetheless, a significantly reduced cone ERG response was observed in RPGRIP1 −/− pet dogs that were clinically normal (Busse et al. 2011), suggesting that the locus mapped in the original research colony could still be pathogenic in the pet population but with a phenotype that was milder or not clinically apparent in some individuals.
To account for the genotype–phenotype discordance in the Japanese pet MLHDs, possible involvement of additional genetic loci was explored. GWAS using 80 RPGRIP1 −/− dogs showing variable phenotypes was performed. A single modifier locus that was homozygous in almost all the early-onset cases was identified and is now under investigation (Miyadera et al. 2012).
It seems likely that the dogs in the UK research colony were all homozygous or “fixed” for the second locus mapped using the Japanese pet dogs. Hence, only by studying the more genetically and clinically heterogeneous pet population could the second locus which modifies the disease onset be identified. In hindsight, the two-step mapping utilizing different subpopulations of MLHD facilitated the identification of two loci involved in cord1.
Dissecting the heterogeneous nature of canine retinal degenerations
Two aspects of genetic heterogeneity among canine RDs have recently emerged. In one situation, breed-specific phenotypes of canine RDs have been shown to arise from the same gene/mutation in different breeds. This is particularly evident in the breed-specific age of onset and rate of progression of the clinical phenotype and is broadly presumed to result from differences of breed-specific “genetic background.” On the other hand, some RDs that are phenotypically similar or even indistinguishable can result from distinct mutations in different genes in different populations. Research in our laboratories and by colleagues elsewhere has shown that both situations occur.
Phenotypic heterogeneity: variable clinical features
Intrafamilial variability has been observed among human RP patients harboring the same mutation (Berson et al. 1991). In contrast to most human populations, dogs have a more uniform genetic background which contributes to consistent disease phenotypes within a breed. However, some canine RD forms are known for substantial phenotypic variability among affected dogs of the same breed.
Among the affected male dogs from the XLPRA1 research colony, extensive variations in the age of onset and disease severity were observed (Zeiss et al. 1999). Because the affected dogs were known to harbor a stable, common mutant RPGR allele, the heterogeneous genetic background due to outcrossing was considered responsible for the phenotypic variation (Zeiss et al. 1999). Haplotypes of six candidate genes known to interact with RPGR or RPGRIP1 were examined for disease-severity association, but no gene-specific haplotypes were found to cosegregate with the moderate or severe phenotype. Also, no significant difference in retinal expression levels of the candidate genes was observed between normal and affected dogs (Guyon et al. 2007). The search for the modifying locus (loci) of XLPRA1 is ongoing.
Significant interbreed variation in the age of onset has been seen among the multiple breeds affected with prcd. The within-breed phenotype is uniform in most affected breeds, and the breed-characteristic age of clinical disease onset ranges from 3 to 12 years (Supplementary Table 2). However, in prcd-affected American Eskimo dog, Nova Scotia duck tolling retriever, Australian cattle dog, and English cocker spaniel, the age of onset varies extensively among PRCD −/− dogs of the same breed. Based on the variable phenotypes observed among affected dogs in a common research facility, genetic rather than environmental factors appear to be the major modifiers.
Variable ages of onset and disease severity has been observed in the ADPRA-affected English mastiffs with a mutation in RHO (Kijas et al. 2002, 2003). In this case, environmental factors such as exposure to light were identified as modifiers, as the mutation that abolished the first consensus glycosylation site rendered the chromophore-free mutant protein extremely unstable (Cideciyan et al. 2005; Gu et al. 2007; Zhu et al. 2004).
Allelic heterogeneity: one gene, different mutations
Different RD forms may arise from different mutations in the same gene, a phenomenon termed allelic heterogeneity. The earliest example in canine RDs is the point mutation and an 8-bp insertion mutation each in exon 21 of PDE6B causing rcd1 and rcd1a, respectively (Dekomien et al. 2000; Suber et al. 1993).
Two distinct forms of X-linked PRA show marked allelic heterogeneity with mutation-specific phenotypes (Zhang et al. 2002). The 5-bp deletion in RPGRORF15 present in XLPRA1 results in a premature stop and truncation of the C-terminus of the protein. The mutation affects only the photoreceptors, and these degenerate in young adults after developing and functioning normally (Zeiss et al. 1999). In contrast, XLPRA2 is an early-onset developmental disease resulting from a 2-bp deletion of RPGRORF15. This deletion causes a frameshift and replacement of a glutamic acid-rich domain by arginine residues before truncation of the C-terminal end of the protein (Zhang et al. 2002). This leads to a toxic gain of function that severely compromises the photoreceptors in the early stages of postnatal development (Beltran et al. 2006).
Allelic heterogeneity is also found in canine achromatopsia and canine multifocal retinopathy. In the former, a 140-kb deletion and a missense mutation in CNGB3 occur in Alaskan Malamutes (cdAMAL), and in German shorthaired pointers (cdGSPT), respectively. The two forms are allelic, and a dog heterozygous for cdAMAL and cdGSPT was found to be achromatopsic (Sidjanin et al. 2002). More recently, distinct breed-specific mutations in the BEST1 gene have been associated with the cmr1, cmr2, and cmr3 forms of canine multifocal retinopathy (Guziewicz et al. 2007; Zangerl et al. 2010).
For some autosomal recessive canine disorders with multiple identified allelic mutations (e.g., rcd1, rcd1a, achromatopsia, and cmr), the phenotype resulting from each allelic series is highly consistent (i.e., regardless of the specific mutant allele), suggesting that the mutations compromise critical functional domains, and the phenotypes represent a loss of function. In contrast, the XLPRA phenotypes represent, in one case, a loss and, in the other case, a toxic gain of function resulting from changes in the size and/or charge of the ORF15 domain, and the phenotypes are markedly different. We would anticipate that yet-to-be-recognized mutations in canine RHO that do not affect consensus glycosylation sites similarly would result in very different phenotypes as in humans (Berson et al. 1991).
Allelism of autosomal recessive forms of early- and late-onset PRA in different breeds has been tested by interbreed crosses. Production of normal progeny from matings among rcd1, rcd2, and erd-affected dogs confirmed nonallelism of these diseases (Acland et al. 1989; Wang et al. 1999). Similarly, nonallelism between erd and prcd (Ray et al. 1996) and rd and prcd (Aguirre 1976) was established. In contrast, allelism for prcd in multiple breeds was demonstrated and was instrumental in LA and positional cloning of the novel retinal disease gene (Aguirre and Acland 1988, 2006).
Genetic heterogeneity: one breed, different forms of RDs
Often, only one form of RD caused by a single mutation segregates in a breed leading to a uniform phenotype. However, in some breeds, more than one form seems to be present (Table 2). Although we suspect that these represent cases of genetic heterogeneity based on molecular testing of known mutations in retinal disease genes, the possibility of allelic heterogeneity cannot be excluded until the causative mutations are identified or time-consuming cross-breeding studies are carried out.
For example, of the more than 22 breeds affected with prcd, Australian cattle dog, Australian stumpy tail cattle dog, Chinese crested, Golden retriever (GR), Labrador retriever, Norwegian elkhound, and Miniature/Toy poodle are now known to each segregate another (non-prcd) form of PRA which remains to be characterized molecularly (Optigen, http://www.optigen.com). In some breeds, DNA tests are currently available for two forms of RDs: examples include prcd and GR_PRA1 in GR, rcd1 and rcd4 in Irish setters, ADPRA and cmr in English mastiff, prcd and osd1 in Labrador retrievers, and XLPRA1 and osd2 in Samoyeds.
Common founder: one disease, multiple breeds
Many canine RDs are breed-specific and each RD is known to segregate in only one breed. However, some RDs are more broadly distributed, caused by a single mutant allele that is identical by descent (IBD). In some cases, breeds segregating an RD allele that is IBD share well-recognized recent common ancestry origin or have experienced interbreed gene flow during breed establishment. In other cases, however, the common ancestry is less obvious whether it is recent or much more ancient.
For example, the BEST1 stop mutation responsible for cmr1 occurs in several Molossus-type (Mastiff-derived) breeds, including the English mastiff, Bullmastiff, Great Pyrenees (Guziewicz et al. 2007), Cane Corso, Dogue de Bordeaux, English and American bulldogs, and Perro de Presa Canario (Guziewicz and Zangerl, personal communication) (Supplementary Fig. 1). Based on historical and morphological relatedness, IBD is easily understood and may represent descent from a relatively recent common ancestor. However, cmr1 phenotypes with the mutation identical to that in the Mastiff breeds was recently identified in the Australian shepherd (Hoffmann, Guziewicz, and Zangerl, personal communication), a breed considered to be genetically distant from the Mastiff breeds (Supplementary Fig. 1). Ongoing studies are exploring further the ancestry and possible gene flow regarding this outlier.
In the case of cea, an identical disease haplotype segregates not only in several Collie-related herding breeds with historic common ancestry, but also in less obviously related nonherding breeds (e.g., Boykin spaniel and Nova Scotia duck tolling retriever), suggesting the possibility of past or recent gene flow from the herding group.
A situation similar to that for cea pertains to canine achromatopsia with an IBD cdAMAL allele segregating in the Alaskan Malamute, Siberian Husky, and Miniature Australian shepherd breeds (Goldstein, personal communication; Yeh and Komaromy, personal communication), suggesting again either a much more ancient origin of the mutant allele or a more recent but unsuspected gene flow between breeds.
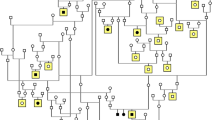
In contrast, the IBD mutant PRCD allele is shared by at least 22 breeds of very diverse origin (Fig. 5). It is likely to be an ancient mutation arising in a common founder before the breeds became isolated breeding populations. This founder mutation is not recognized to segregate in the cluster of “ancient” breeds that trace their ancestry to Asia or Africa (e.g., Akita, Chinese shar-pei, Siberian Husky, Basenji, or Afghan hound), emphasizing the distinctly more ancient origin of these breeds (Parker et al. 2004; Vonholdt et al. 2010).

Diverse breeds share a common identical-by-descent mutation in PRCD. Genetic diversity of 17 of 22 dog breeds that segregate the C2Y mutation in PRCD is shown in a neighbor-joining tree of domestic dogs and gray wolves (reprinted and modified with permission from Vonholdt et al. 2010). The names of breeds that were included in the study by Vonholdt et al. (2010) are outlined. For other breeds, images are placed at a hypothetical position based on breed history and previously reported structural analysis of canine breeds (Parker et al. 2004). Dog images are from Wikipedia (http://en.wikipedia.org/) and not to scale
Phenocopies: nongenetic retinopathies
Acquired forms of RDs such as sudden acquired retinal degeneration (SARD) may complicate the diagnosis and ascertainment of RDs (Acland et al. 1984; Cullen and Grahn 2002; Keller et al. 2006; Miller et al. 1998; O’Toole et al. 1992; Vainisi et al. 1983; van der Woerdt et al. 1991). SARD appears clinically similar to PRA/CRD except that the onset of total blindness is sudden whereas the vision loss is progressive in PRA/CRD. To clearly differentiate SARD from PRA/CRD, it is essential that examination be done at the onset of blindness by ophthalmoscopy (normal fundus) and ERG testing (nonrecordable ERG). At the end stage, both disorders are indistinguishable.
DNA testing
Almost all mutations that underlie RDs can be screened for by tests that examine a DNA sample for the presence or the absence of an RD mutation known or suspected to be segregating in the respective breed. Because of extensive genetic and allelic heterogeneities, molecular diagnoses of sporadic human RD patients requires screening of hundreds of genes and thousands of mutations, often using microarray chips (Stone 2007; Zernant et al. 2005). In contrast, DNA testing of only the known mutation(s) associated with the respective canine breed is often sufficient for the molecular diagnosis of RDs.
A DNA test has advantages over other clinical diagnostic methods in that it can detect unaffected carriers as well as subclinical cases prior to the onset of disease. This allows selection of healthy reproductive pairs suitable for breeding. However, if there are other unidentified mutations causing other forms of RD in the breed, the currently available DNA test alone will not be able to exclude all forms of inherited RD in the breed.
Even when there is good correlation between the DNA test result and the retinal phenotype, a broader perspective is needed when applying the DNA test results to breeding programs. Nonretinal traits that cannot yet be tested for molecularly should also be taken into account to assess the overall health and genetic potential of the dog. Conservation of the within-breed genetic diversity also requires consideration when there is selective pressure by DNA testing, particularly if the mutation frequency is high. A sound breeding strategy will aim to prevent producing affected offspring and more gradually eliminate the RD mutant allele from the population, while at the same time maintaining the genetic diversity of a breed.
Prospects of canine retinal disease research
Study population
More than a dozen of canine RD research colonies have been developed for mapping, phenotype characterization, investigation of the disease pathology, and for gene or other therapies. While successful mapping of many diseases has used purpose-bred research colonies, some studies benefitted from recruiting cases and controls from client-owned pet dogs. Using samples and clinical data from client-owned dogs, the chromosomal location of prcd in the American Eskimo and Finish Lapphund breeds was independently mapped by homozygosity mapping using only 13 (Moody et al. 2005) and 12 (Aguirre-Hernandez et al. 2007) affected cases, respectively. The use of pet dogs eliminates the difficulties and expense of developing and maintaining a research colony. Sporadic cases from the pet population are ideal for case–control association studies as unrelated cases and controls will be studied without requiring prior assumptions about the mode of inheritance.
The drawbacks of studying pet dogs are the difficulties of collecting enough cases and suitable controls, particularly when the mutant allele frequency in the population is low. Also, the use of pet dogs may not be ideal for LA mapping where sufficiently large and informative pedigrees are required. Furthermore, as consistent diagnostic criteria are crucial for distinguishing different forms of RDs, standardization of phenotyping remains a challenge. Our experience in searching for the prcd locus defect indicated that approximately one third of the clinical records received were incomplete or inaccurate and could not be used in the homozygosity mapping studies used to reduce the critical interval. This problem is more serious when dealing with late- versus early-onset diseases because acquired retinal abnormalities are more common in older dogs and are a confounding variable.
Provided that rigorous phenotype ascertainment is obtained, the study population for GWAS can originate from the general pet population segregating the trait of interest, provided that there is sufficient cooperation of the owners, breeders, and referring veterinary clinicians. On the other hand, research colonies continue to play a critical role in providing adequate tissue resources to functionally evaluate positional candidate genes or changes in the DNA sequence.
Man to dog—dog to man
At least 24 mutations in 18 genes underlying canine RDs have been identified to date (Table 1). If we extrapolate from the human disease data, there may be more than 214 genes and loci critical for retinal function (Fig. 1) (Supplementary Table 1). While mutations in some of the human RD genes may not be associated with a recognizable phenotype in dogs (e.g., mutations in cone opsin genes that cause color vision deficits in humans without affecting visual acuity) (Nathans et al. 1986a, b), mutations in the majority of these genes would be expected to be associated with a recognizable phenotype in dogs.
On the other hand, genetic and pathologic findings derived from studies of canine RDs have contributed to the understanding of human disease. PRCD associated with prcd in multiple breeds was a novel gene first identified in dogs (Zangerl et al. 2006) and subsequently in humans. Strikingly, this led to the identification of an RP patient homozygous for the same G5A (C2Y) mutation that causes prcd in dogs, as well as other mutations in this gene (Nevet et al. 2010). Other examples of RD genes first described in the dog are CCDC66 and SLC4A3 underlying gPRASPD (Dekomien et al. 2010) and GR_PRA1 (Downs et al. 2011), respectively. Both loci were mapped by GWAS without presumption of the genes involved. CCDC66 was subsequently disrupted in a mouse model whose progressive rod–cone dysplasia phenotype reinforced the finding in the Schapendoes dog (Gerding et al. 2011).
Gene therapy
Identification of the genes and mutations underlying canine RDs have provided an excellent testing ground for gene replacement therapy, one of the approaches under active development to prevent, stabilize, or reverse the retinal degenerative process in man and animals.
Naturally occurring cLCA in Briards with a mutation in RPE65 has been treated successfully with subretinal injection of a recombinant adeno-associated virus 2 carrying wild-type RPE65 (Acland et al. 2001; Narfström et al. 2003). The single injection was safe, effective, and stable, with functional improvement up to 4 years post injection (Acland et al. 2005; Narfström et al. 2008). The treatment was subsequently applied to human LCA patients carrying RPE65 mutations and several trials have achieved significant restoration of visual function (Bainbridge et al. 2008; Hauswirth et al. 2008; Maguire et al. 2008).
Successful gene therapy using the dog model has also been achieved in CNGB3 achromatopsia (cdAMAL, cdGSPT) (Komaromy et al. 2010). Therapy directed specifically to cone photoreceptors led to the restoration of ERG cone function, day vision, and preservation of structure. Treatment was stable for the nearly 3-year study duration.
Conclusion
Since the first identification of the PDE6B mutation in 1993, there has been significant progress in the molecular investigation of canine RDs, benefiting from the rich genetic resources and tools developed over the years. Completion of the canine reference genome in 2005 further revolutionized the process of searching for genes and mutations underlying these and other diseases in dogs.
Canine RDs are true disease homologs for specific human disorders based on the phenotypic similarity and the common underlying gene defects, providing excellent models for analysis of the molecular mechanisms of these disorders. The number of canine RDs characterized at the molecular level will increase even further over the coming years, with GWAS being the current tool of choice and with rapid cost-effective whole-genome sequencing looming on the horizon.
Canine RDs have also been developed as an excellent model system to develop and test novel therapies applicable to humans. Because of the similarities in disease phenotype and the similar structure and function of the canine and human retinas, dogs are ideally suited for developing these therapies. Thus, the transition from gene discovery to gene therapy leading to the application to human medicine has been made possible.
The unique phenotypic and genetic uniformity as well as variability within and across dog breeds has been the key to the dramatic progress in the molecular understanding of RDs in dogs and will continue to be helpful in discovering retinal and other diseases in the future.
References
Abramov I, Gordon J, Hendrickson A, Hainline L, Dobson V, LaBossiere E (1982) The retina of the newborn human infant. Science 217:265–267
Acland GM, Aguirre GD (1987) Retinal degenerations in the dog: IV. Early retinal degeneration (erd) in Norwegian elkhounds. Exp Eye Res 44:491–521
Acland GM, Aguirre GD (1995) Oculoskeletal dysplasias in Samoyed and Labrador retriever dogs: nonallelic disorders akin to Stickler-like syndromes affecting humans. In: 2nd international DOGMAP meeting, Cambridge
Acland GM, Irby NL, Aguirre GD, Gross S (1984) Sudden acquired retinal degeneration in the dog: clinical and morphologic characterization of the “silent retina” syndrome. Trans Am Coll Vet Ophthalmol 15:86–104
Acland G, Fletcher R, Gentleman S, Chader G, Aguirre G (1989) Non-allelism of three genes (rcd1, rcd2, erd) for early-onset hereditary retinal degeneration. Exp Eye Res 49:983–998
Acland GM, Blanton SH, Hershfield B, Aguirre GD (1994) XLPRA: a canine retinal degeneration inherited as an X-linked trait. Am J Med Genet 52:27–33
Acland GM, Ray K, Mellersh CS, Gu W, Langston AA, Rine J, Ostrander EA, Aguirre GD (1998) Linkage analysis and comparative mapping of canine progressive rod–cone degeneration (prcd) establishes potential locus homology with retinitis pigmentosa (RP17) in humans. Proc Natl Acad Sci USA 95:3048–3053
Acland GM, Ray K, Mellersh CS, Langston AA, Rine J, Ostrander EA, Aguirre GD (1999) A novel retinal degeneration locus identified by linkage and comparative mapping of canine early retinal degeneration. Genomics 59:134–142
Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, Cideciyan AV, Pearce-Kelling SE, Anand V, Zeng Y, Maguire AM, Jacobson SG, Hauswirth WW, Bennett J (2001) Gene therapy restores vision in a canine model of childhood blindness. Nat Genet 28:92–95
Acland GM, Aguirre GD, Bennett J, Aleman TS, Cideciyan AV, Bennicelli J, Dejneka NS, Pearce-Kelling SE, Maguire AM, Palczewski K, Hauswirth WW, Jacobson SG (2005) Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther 12:1072–1082
Aguirre GD (1976) Inherited retinal degenerations in the dog. Trans Am Acad Ophthalmol Otolaryngol 81:667–676
Aguirre G (1978) Retinal degenerations in the dog. I. Rod dysplasia. Exp Eye Res 26:233–253
Aguirre GD, Acland GM (1988) Variation in retinal degeneration phenotype inherited at the prcd locus. Exp Eye Res 46:663–687
Aguirre GD, Acland GM (2006) Models, mutants and man: searching for unique phenotypes and genes in the dog model of inherited retinal degeneration. In: Ostrander EA, Giger U, Lindblad-Toh K (eds) The dog and its genome. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, pp 291–325
Aguirre GD, Rubin LF (1974) Pathology of hemeralopia in the Alaskan malamute dog. Invest Ophthalmol 13:231–235
Aguirre GD, Rubin LF (1975) The electroretinogram in dogs with inherited cone degeneration. Invest Ophthalmol 14:840–847
Aguirre GD, Rubin LF, Bistner SI (1972) Development of the canine eye. Am J Vet Res 33:2399–2414
Aguirre GD, Lolley R, Farber D, Fletcher T, Chader GJ (1978) Rod–cone dysplasia in Irish setter dogs: a defect in cyclic GMP metabolism in visual cells. Science 201:1133–1135
Aguirre G, Alligood J, O’Brien P, Buyukmihci N (1982a) Pathogenesis of progressive rod–cone degeneration in miniature poodles. Invest Ophthalmol Vis Sci 23:610–630
Aguirre G, Farber D, Lolley R, O’Brien P, Alligood J, Fletcher RT, Chader G (1982b) Retinal degeneration in the dog. III. Abnormal cyclic nucleotide metabolism in rod–cone dysplasia. Exp Eye Res 35:625–642
Aguirre GD, Baldwin V, Pearce-Kelling S, Narfstrom K, Ray K, Acland GM (1998) Congenital stationary night blindness in the dog: common mutation in the RPE65 gene indicates founder effect. Mol Vis 4:23–29
Aguirre-Hernandez J, Sargan DR (2005) Evaluation of candidate genes in the absence of positional information: a poor bet on a blind dog! J Hered 96:475–484
Aguirre-Hernandez J, Wickstrom K, Sargan DR (2007) The Finnish lapphund retinal atrophy locus maps to the centromeric region of CFA9. BMC Vet Res 3:14
Andersson L (2009) Genome-wide association analysis in domestic animals: a powerful approach for genetic dissection of trait loci. Genetica 136:341–349
Awano T, Johnson GS, Wade CM, Katz ML, Johnson GC, Taylor JF, Perloski M, Biagi T, Baranowska I, Long S, March PA, Olby NJ, Shelton GD, Khan S, O’Brien DP, Lindblad-Toh K, Coates JR (2009) Genome-wide association analysis reveals a SOD1 mutation in canine degenerative myelopathy that resembles amyotrophic lateral sclerosis. Proc Natl Acad Sci USA 106:2794–2799
Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, Viswanathan A, Holder GE, Stockman A, Tyler N, Petersen-Jones S, Bhattacharya SS, Thrasher AJ, Fitzke FW, Carter BJ, Rubin GS, Moore AT, Ali RR (2008) Effect of gene therapy on visual function in Leber’s congenital amaurosis. N Engl J Med 358:2231–2239
Bannasch D, Young A, Myers J, Truve K, Dickinson P, Gregg J, Davis R, Bongcam-Rudloff E, Webster MT, Lindblad-Toh K, Pedersen N (2010) Localization of canine brachycephaly using an across breed mapping approach. PLoS One 5:e9632
Barnett K (1965) Canine retinopathies–III. The other breeds. J Small Anim Pract 6:185–196
Barnett K, Stades F (1979) Collie eye anomaly in the Shetland sheepdog in the Netherlands. J Small Anim Pract 20:321–329
Barnett K, Bjorck G, Kock E (1970) Hereditary retinal dysplasia in the labrador retriever in England and in Sweden. J Small Anim Pract 10:755–759
Beltran WA, Hammond P, Acland GM, Aguirre GD (2006) A frameshift mutation in RPGR exon ORF15 causes photoreceptor degeneration and inner retina remodeling in a model of X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci 47:1669–1681
Berson E, Rosner B, Sandberg M, Dryja T (1991) Ocular findings in patients with autosomal dominant retinitis pigmentosa and a rhodopsin gene defect (PRO-23-HIS). Arch Ophthalmol 109:92–101
Booij JC, Florijn RJ, ten Brink JB, Loves W, Meire F, van Schooneveld MJ, de Jong PT, Bergen AA (2005) Identification of mutations in the AIPL1, CRB1, GUCY2D, RPE65, and RPGRIP1 genes in patients with juvenile retinitis pigmentosa. J Med Genet 42:e67
Bowes C, Li T, Danciger M, Baxter LC, Applebury ML, Farber DB (1990) Retinal degeneration in the rd mouse is caused by a defect in the ß subunit of rod cGMP-phosphodiesterase. Nature 347:677–680
Boyko AR, Quignon P, Li L, Schoenebeck JJ, Degenhardt JD, Lohmueller KE, Zhao K, Brisbin A, Parker HG, von Holdt BM, Cargill M, Auton A, Reynolds A, Elkahloun AG, Castelhano M, Mosher DS, Sutter NB, Johnson GS, Novembre J, Hubisz MJ, Siepel A, Wayne RK, Bustamante CD, Ostrander EA (2010) A simple genetic architecture underlies morphological variation in dogs. PLoS Biol 8:e1000451
Breen M, Jouquand S, Renier C, Mellersh CS, Hitte C, Holmes NG, Chéron A, Suter N, Vignaux F, Bristow AE, Priat C, McCann E, André C, Boundy S, Gitsham P, Thomas R, Bridge WL, Spriggs HF, Ryder EJ, Curson A, Sampson J, Ostrander EA, Binns MM, Galibert F (2001) Chromosome-specific single-locus FISH probes allow anchorage of an 1800-marker integrated radiation-hybrid/linkage map of the domestic dog genome to all chromosomes. Genome Res 11:1784–1795
Busse C, Barnett KC, Mellersh CS, Adams VJ (2011) Ophthalmic and cone derived electrodiagnostic findings in outbred miniature long-haired dachshunds homozygous for a RPGRIP1 mutation. Vet Ophthalmol 14:146–152
Cadieu E, Neff MW, Quignon P, Walsh K, Chase K, Parker HG, Vonholdt BM, Rhue A, Boyko A, Byers A, Wong A, Mosher DS, Elkahloun AG, Spady TC, Andre C, Lark KG, Cargill M, Bustamante CD, Wayne RK, Ostrander EA (2009) Coat variation in the domestic dog is governed by variants in three genes. Science 326:150–153
Carrig C, Sponenberg D, Schmidt G, Tvedten H (1988) Inheritance of associated ocular and skeletal dysplasia in Labrador retrievers. J Am Vet Med Assoc 193:1269–1272
Cideciyan AV, Jacobson SG, Aleman TS, Gu D, Pearce-Kelling SE, Sumaroka A, Acland GM, Aguirre GD (2005) In vivo dynamics of retinal injury and repair in the rhodopsin mutant dog model of human retinitis pigmentosa. Proc Natl Acad Sci USA 102:5233–5238
Clements PJ, Gregory CY, Peterson-Jones SM, Sargan DR, Bhattacharya SS (1993) Confirmation of the rod cGMP phosphodiesterase ß subunit (PDEß) nonsense mutation in affected rcd-1 Irish setters in the UK and development of a diagnostic test. Curr Eye Res 12:861–866
Cullen CL, Grahn BH (2002) Diagnostic ophthalmology. Acute prechiasmal blindness due to sudden acquired retinal degeneration syndrome. Can Vet J 43:729–730
Curcio CA, Sloan KR, Kalina RE, Hendrickson AE (1990) Human photoreceptor topography. J Comp Neurol 292:497–523
Curtis R, Barnett KC (1993) Progressive retinal atrophy in miniature longhaired dachshund dogs. Br Vet J 149:71–85
Dekomien G, Epplen JT (2002a) The canine Phosducin gene: characterization of the exon-intron structure and exclusion as a candidate gene for generalized progressive retinal atrophy in 11 dog breeds. Mol Vis 8:138–142
Dekomien G, Epplen JT (2002b) The canine recoverin (RCV1) gene: a candidate gene for generalized progressive retinal atrophy. Mol Vis 8:436–441
Dekomien G, Epplen JT (2002c) Screening of the arrestin gene in dogs afflicted with generalized progressive retinal atrophy. BMC Genet 3:12
Dekomien G, Epplen JT (2003) Analysis of PDE6D and PDE6G genes for generalised progressive retinal atrophy (gPRA) mutations in dogs. Genet Sel Evol 35:445–456
Dekomien G, Runte M, Godde R, Epplen JT (2000) Generalized progressive retinal atrophy of Sloughi dogs is due to an 8-bp insertion in exon 21 of the PDE6B gene. Cytogenet Cell Genet 90:261–267
Dekomien G, Vollrath C, Petrasch-Parwez E, Boeve MH, Akkad DA, Gerding WM, Epplen JT (2010) Progressive retinal atrophy in Schapendoes dogs: mutation of the newly identified CCDC66 gene. Neurogenetics 11:163–174
Dodman NH, Karlsson EK, Moon-Fanelli A, Galdzicka M, Perloski M, Shuster L, Lindblad-Toh K, Ginns EI (2010) A canine chromosome 7 locus confers compulsive disorder susceptibility. Mol Psychiatry 15:8–10
Downs LM, Wallin-Hakansson B, Boursnell M, Marklund S, Hedhammar A, Truve K, Hubinette L, Lindblad-Toh K, Bergstrom T, Mellersh CS (2011) A frameshift mutation in Golden Retriever dogs with progressive retinal atrophy endorses SLC4A3 as a candidate gene for human retinal degenerations. PLoS One 6:e21452
Drogemuller C, Karlsson EK, Hytonen MK, Perloski M, Dolf G, Sainio K, Lohi H, Lindblad-Toh K, Leeb T (2008) A mutation in hairless dogs implicates FOXI3 in ectodermal development. Science 321:1462
Drogemuller C, Becker D, Brunner A, Haase B, Kircher P, Seeliger F, Fehr M, Baumann U, Lindblad-Toh K, Leeb T (2009) A missense mutation in the SERPINH1 gene in dachshunds with osteogenesis imperfecta. PLoS Genet 5:e1000579
Drogemuller C, Becker D, Kessler B, Kemter E, Tetens J, Jurina K, Jaderlund KH, Flagstad A, Perloski M, Lindblad-Toh K, Matiasek K (2010) A deletion in the N-myc downstream regulated gene 1 (NDRG1) gene in Greyhounds with polyneuropathy. PLoS One 5:e11258
Dryja TP, Adams SM, Grimsby JL, McGee TL, Hong DH, Li T, Andreasson S, Berson EL (2001) Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am J Hum Genet 68:1295–1298
Farber D, Lolley R (1974) Cyclic guanosine monophosphate: elevation in degenerating photoreceptor cells of the C3H mouse retina. Science 186:449–451
Farber DB, Danciger JS, Aguirre G (1992) The beta subunit of cyclic GMP phosphodiesterase mRNA is deficient in canine rod–cone dysplasia 1. Neuron 9:349–356
Farias FH, Zeng R, Johnson GS, Wininger FA, Taylor JF, Schnabel RD, McKay SD, Sanders DN, Lohi H, Seppala EH, Wade CM, Lindblad-Toh K, O’Brien DP, Katz ML (2011) A truncating mutation in ATP13A2 is responsible for adult-onset neuronal ceroid lipofuscinosis in Tibetan terriers. Neurobiol Dis 42:468–474
Farrar GJ, McWilliam P, Bradley DG, Kenna P, Lawler M, Sharp EM, Humphries MM, Eiberg H, Conneally PM, Trofatter JA et al (1990) Autosomal dominant retinitis pigmentosa: linkage to rhodopsin and evidence for genetic heterogeneity. Genomics 8:35–40
Gerber S, Perrault I, Hanein S, Barbet F, Ducroq D, Ghazi I, Martin-Coignard D, Leowski C, Homfray T, Dufier JL, Munnich A, Kaplan J, Rozet JM (2001) Complete exon-intron structure of the RPGR-interacting protein (RPGRIP1) gene allows the identification of mutations underlying Leber congenital amaurosis. Eur J Hum Genet 9:561–571
Gerding WM, Schreiber S, Schulte-Middelmann T, de Castro Marques A, Atorf J, Akkad DA, Dekomien G, Kremers J, Dermietzel R, Gal A, Rulicke T, Ibrahim S, Epplen JT, Petrasch-Parwez E (2011) Ccdc66 null mutation causes retinal degeneration and dysfunction. Hum Mol Genet 20(18):3620–3631
Goldstein O, Zangerl B, Pearce-Kelling S, Sidjanin DJ, Kijas JW, Felix J, Acland GM, Aguirre GD (2006) Linkage disequilibrium mapping in domestic dog breeds narrows the progressive rod–cone degeneration interval and identifies ancestral disease-transmitting chromosome. Genomics 88:541–550
Goldstein O, Guyon R, Kukekova A, Kuznetsova TN, Pearce-Kelling SE, Johnson J, Aguirre GD, Acland GM (2010a) COL9A2 and COL9A3 mutations in canine autosomal recessive oculoskeletal dysplasia. Mamm Genome 21:398–408
Goldstein O, Kukekova AV, Aguirre GD, Acland GM (2010b) Exonic SINE insertion in STK38L causes canine early retinal degeneration (erd). Genomics 96:362–368
Goldstein O, Mezey JG, Boyko AR, Gao C, Wang W, Bustamante CD, Anguish LJ, Jordan JA, Pearce-Kelling SE, Aguirre GD, Acland GM (2010c) An ADAM9 mutation in canine cone–rod dystrophy 3 establishes homology with human cone–rod dystrophy 9. Mol Vis 16:1549–1569
Gould DJ, Petersen-Jones SM, Sohal A, Barnett KC, Sargan DR (1995) Investigation of the role of opsin gene polymorphism in generalized progressive retinal atrophies in dogs. Anim Genet 26:261–267
Gould DJ, Petersen-Jones SM, Lin CT, Sargan DR (1997) Cloning of canine rom-1 and its investigation as a candidate gene for generalized progressive retinal atrophies in dogs. Anim Genet 28:391–396
Gray MM, Granka JM, Bustamante CD, Sutter NB, Boyko AR, Zhu L, Ostrander EA, Wayne RK (2009) Linkage disequilibrium and demographic history of wild and domestic canids. Genetics 181:1493–1505
Gu D, Beltran WA, Li Z, Acland GM, Aguirre GD (2007) Clinical light exposure, photoreceptor degeneration, and AP-1 activation: a cell death or cell survival signal in the rhodopsin mutant retina? Invest Ophthalmol Vis Sci 48:4907–4918
Guyon R, Lorentzen TD, Hitte C, Kim L, Cadieu E, Parker HG, Quignon P, Lowe JK, Renier C, Gelfenbeyn B, Vignaux F, DeFrance HB, Gloux S, Mahairas GG, André C, Galibert F, Ostrander EA (2003) A 1-Mb resolution radiation hybrid map of the canine genome. Proc Natl Acad Sci USA 100:5296–5301
Guyon R, Pearce-Kelling SE, Zeiss CJ, Acland GM, Aguirre GD (2007) Analysis of six candidate genes as potential modifiers of disease expression in canine XLPRA1, a model for human X-linked retinitis pigmentosa 3. Mol Vis 13:1094–1105
Guziewicz KE, Zangerl B, Lindauer SJ, Mullins RF, Sandmeyer LS, Grahn BH, Stone EM, Acland GM, Aguirre GD (2007) Bestrophin gene mutations cause canine multifocal retinopathy: a novel animal model for best disease. Invest Ophthalmol Vis Sci 48:1959–1967
Hameed A, Abid A, Aziz A, Ismail M, Mehdi SQ, Khaliq S (2003) Evidence of RPGRIP1 gene mutations associated with recessive cone–rod dystrophy. J Med Genet 40:616–619
Hauswirth WW, Aleman TS, Kaushal S, Cideciyan AV, Schwartz SB, Wang L, Conlon TJ, Boye SL, Flotte TR, Byrne BJ, Jacobson SG (2008) Treatment of Leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther 19:979–990
Hunt DM, Buch P, Michaelides M (2010) Guanylate cyclases and associated activator proteins in retinal disease. Mol Cell Biochem 334:157–168
Karlsson EK, Baranowska I, Wade CM, Salmon Hillbertz NH, Zody MC, Anderson N, Biagi TM, Patterson N, Pielberg GR, Kulbokas EJ 3rd, Comstock KE, Keller ET, Mesirov JP, von Euler H, Kampe O, Hedhammar A, Lander ES, Andersson G, Andersson L, Lindblad-Toh K (2007) Efficient mapping of Mendelian traits in dogs through genome-wide association. Nat Genet 39:1321–1328
Karlstam L, Hertil E, Zeiss C, Ropstad EO, Bjerkas E, Dubielzig RR, Ekesten B (2011) A slowly progressive retinopathy in the Shetland Sheepdog. Vet Ophthalmol 14:227–238
Keller RL, Kania SA, Hendrix DV, Ward DA, Abrams K (2006) Evaluation of canine serum for the presence of antiretinal autoantibodies in sudden acquired retinal degeneration syndrome. Vet Ophthalmol 9:195–200
Kijas JW, Cideciyan AV, Aleman TS, Pianta MJ, Pearce-Kelling SE, Miller BJ, Jacobson SG, Aguirre GD, Acland GM (2002) Naturally occurring rhodopsin mutation in the dog causes retinal dysfunction and degeneration mimicking human dominant retinitis pigmentosa. Proc Natl Acad Sci USA 99:6328–6333
Kijas JW, Miller BJ, Pearce-Kelling SE, Aguirre GD, Acland GM (2003) Canine models of ocular disease: outcross breedings define a dominant disorder present in the English mastiff and bull mastiff dog breeds. J Hered 94:27–30
Kijas JW, Zangerl B, Miller B, Nelson J, Kirkness EF, Aguirre GD, Acland GM (2004) Cloning of the canine ABCA4 gene and evaluation in canine cone–rod dystrophies and progressive retinal atrophies. Mol Vis 10:223–232
Kirkness EF, Bafna V, Halpern AL, Levy S, Remington K, Rusch DB, Delcher AL, Pop M, Wang W, Fraser CM, Venter JC (2003) The dog genome: survey sequencing and comparative analysis. Science 301:1898–1903
Klein W, Dekomien G, Holmes N, Epplen JT (1998) Evaluation of ROM1 as a candidate gene in generalised progressive retinal atrophy in dogs. Anim Genet 29:316–318
Komaromy AM, Alexander JJ, Rowlan JS, Garcia MM, Chiodo VA, Kaya A, Tanaka JC, Acland GM, Hauswirth WW, Aguirre GD (2010) Gene therapy rescues cone function in congenital achromatopsia. Hum Mol Genet 19:2581–2593
Kropatsch R, Petrasch-Parwez E, Seelow D, Schlichting A, Gerding WM, Akkad DA, Epplen JT, Dekomien G (2010) Generalized progressive retinal atrophy in the Irish Glen of Imaal Terrier is associated with a deletion in the ADAM9 gene. Mol Cell Probes 24(6):357–363
Kuchtey J, Olson LM, Rinkoski T, Mackay EO, Iverson TM, Gelatt KN, Haines JL, Kuchtey RW (2011) Mapping of the disease locus and identification of ADAMTS10 as a candidate gene in a canine model of primary open angle glaucoma. PLoS Genet 7:e1001306
Kukekova AV, Nelson J, Kuchtey RW, Lowe JK, Johnson JL, Ostrander EA, Aguirre GD, Acland GM (2006) Linkage mapping of canine rod cone dysplasia type 2 (rcd2) to CFA7, the canine orthologue of human 1q32. Invest Ophthalmol Vis Sci 47:1210–1215
Kukekova AV, Goldstein O, Johnson JL, Richardson MA, Pearce-Kelling SE, Swaroop A, Friedman JS, Aguirre GD, Acland GM (2009) Canine RD3 mutation establishes rod–cone dysplasia type 2 (rcd2) as ortholog of human and murine rd3. Mamm Genome 20:109–123
Langston AA, Mellersh CS, Neal CL, Ray K, Acland GM, Gibbs M, Aguirre GD, Fournier RE, Ostrander EA (1997) Construction of a panel of canine-rodent hybrid cell lines for use in partitioning of the canine genome. Genomics 46:317–325
Leskov IB, Klenchin VA, Handy JW, Whitlock GG, Govardovskii VI, Bownds MD, Lamb TD, Pugh EN Jr, Arshavsky VY (2000) The gain of rod phototransduction: reconciliation of biochemical and electrophysiological measurements. Neuron 27:525–537
Li R, Mignot E, Faraco J, Kadotani H, Cantanese J, Zhao B, Lin X, Hinton L, Ostrander EA, Patterson DF, de Jong PJ (1999) Construction and characterization of an eightfold redundant dog genomic bacterial artificial chromosome library. Genomics 58:9–17
Lin CT, Petersen-Jones SM, Sargan DR (1998) Isolation and investigation of canine phosducin as a candidate for canine generalized progressive retinal atrophies. Exp Eye Res 67:473–480
Lindblad-Toh K, Wade CM, Mikkelsen TS, Karlsson EK, Jaffe DB, Kamal M, Clamp M, Chang JL, Kulbokas EJ, Zody MC, Mauceli E, Xie X, Breen M, Wayne RK, Ostrander EA, Ponting CP, Galibert F, Smith DR, Dejong PJ, Kirkness E, Alvarez P, Biagi T, Brockman W, Butler J, Chin CW, Cook A, Cuff J, Daly MJ, Decaprio D, Gnerre S, Grabherr M, Kellis M, Kleber M, Bardeleben C, Goodstadt L, Heger A, Hitte C, Kim L, Koepfli KP, Parker HG, Pollinger JP, Searle SM, Sutter NB, Thomas R, Webber C, Baldwin J, Abebe A, Abouelleil A, Aftuck L, Ait-Zahra M, Aldredge T, Allen N, An P, Anderson S, Antoine C, Arachchi H, Aslam A, Ayotte L, Bachantsang P, Barry A, Bayul T, Benamara M, Berlin A, Bessette D, Blitshteyn B, Bloom T, Blye J, Boguslavskiy L, Bonnet C, Boukhgalter B, Brown A, Cahill P, Calixte N, Camarata J, Cheshatsang Y, Chu J, Citroen M, Collymore A, Cooke P, Dawoe T, Daza R, Decktor K, Degray S, Dhargay N, Dooley K, Dooley K, Dorje P, Dorjee K, Dorris L, Duffey N, Dupes A, Egbiremolen O, Elong R, Falk J, Farina A, Faro S, Ferguson D, Ferreira P, Fisher S, Fitzgerald M, Foley K, Foley C, Franke A, Friedrich D, Gage D, Garber M, Gearin G, Giannoukos G, Goode T, Goyette A, Graham J, Grandbois E, Gyaltsen K, Hafez N, Hagopian D, Hagos B, Hall J, Healy C, Hegarty R, Honan T, Horn A, Houde N, Hughes L, Hunnicutt L, Husby M, Jester B, Jones C, Kamat A, Kanga B, Kells C, Khazanovich D, Kieu AC, Kisner P, Kumar M, Lance K, Landers T, Lara M, Lee W, Leger JP, Lennon N, Leuper L, Levine S, Liu J, Liu X, Lokyitsang Y, Lokyitsang T, Lui A, Macdonald J, Major J, Marabella R, Maru K, Matthews C, McDonough S, Mehta T, Meldrim J, Melnikov A, Meneus L, Mihalev A, Mihova T, Miller K, Mittelman R, Mlenga V, Mulrain L, Munson G, Navidi A, Naylor J, Nguyen T, Nguyen N, Nguyen C, Nguyen T, Nicol R, Norbu N, Norbu C, Novod N, Nyima T, Olandt P, O’Neill B, O’Neill K, Osman S, Oyono L, Patti C, Perrin D, Phunkhang P, Pierre F, Priest M, Rachupka A, Raghuraman S, Rameau R, Ray V, Raymond C, Rege F, Rise C, Rogers J, Rogov P, Sahalie J, Settipalli S, Sharpe T, Shea T, Sheehan M, Sherpa N, Shi J, Shih D, Sloan J, Smith C, Sparrow T, Stalker J, Stange-Thomann N, Stavropoulos S, Stone C, Stone S, Sykes S, Tchuinga P, Tenzing P, Tesfaye S, Thoulutsang D, Thoulutsang Y, Topham K, Topping I, Tsamla T, Vassiliev H, Venkataraman V, Vo A, Wangchuk T, Wangdi T, Weiand M, Wilkinson J, Wilson A, Yadav S, Yang S, Yang X, Young G, Yu Q, Zainoun J, Zembek L, Zimmer A, Lander ES (2005) Genome sequence, comparative analysis and haplotype structure of the domestic dog. Nature 438:803–819
Lippmann T, Jonkisz A, Dobosz T, Petrasch-Parwez E, Epplen JT, Dekomien G (2007) Haplotype-defined linkage region for gPRA in Schapendoes dogs. Mol Vis 13:174–180
Lowe JK, Kukekova AV, Kirkness EF, Langlois MC, Aguirre GD, Acland GM, Ostrander EA (2003) Linkage mapping of the primary disease locus for collie eye anomaly. Genomics 82:86–95
Magnusson H (1909) On night blindness in the dog following inbreeding. Svensk Vet Tidskr 14:462–466
Magnusson H (1910) Retinitis pigmentosa and consanguinity in the dog. Svensk Vet Tidskr 15:378–380
Magnusson H (1911) Über retinitis pigmentosa und Konsanguinität beim hunde. Arch Vergleichende Ophthal 2:147–163
Magnusson H (1917) Noch ein fall von nachtblindheit beim hunde. Graefe’s Arch Ophthal 93:404–411
Maguire AM, Simonelli F, Pierce EA, Pugh EN Jr, Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, Rossi S, Lyubarsky A, Arruda VR, Konkle B, Stone E, Sun J, Jacobs J, Dell’Osso L, Hertle R, Ma JX, Redmond TM, Zhu X, Hauck B, Zelenaia O, Shindler KS, Maguire MG, Wright JF, Volpe NJ, McDonnell JW, Auricchio A, High KA, Bennett J (2008) Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med 358:2240–2248
McWilliam P, Farrar GJ, Kenna P, Bradley DG, Humphries MM, Sharp EM, McConnell DJ, Lawler M, Sheils D, Ryan C et al (1989) Autosomal dominant retinitis pigmentosa (ADRP): localization of an ADRP gene to the long arm of chromosome 3. Genomics 5:619–622
Mellersh CS, Langston AA, Acland GM, Fleming MA, Ray K, Wiegand NA, Francisco LV, Gibbs M, Aguirre GD, Ostrander EA (1997) A linkage map of the canine genome. Genomics 46:326–336
Mellersh CS, Boursnell ME, Pettitt L, Ryder EJ, Holmes NG, Grafham D, Forman OP, Sampson J, Barnett KC, Blanton S, Binns MM, Vaudin M (2006) Canine RPGRIP1 mutation establishes cone–rod dystrophy in miniature longhaired dachshunds as a homologue of human Leber congenital amaurosis. Genomics 88:293–301
Meurs KM, Mauceli E, Lahmers S, Acland GM, White SN, Lindblad-Toh K (2010) Genome-wide association identifies a deletion in the 3′ untranslated region of striatin in a canine model of arrhythmogenic right ventricular cardiomyopathy. Hum Genet 128:315–324
Meyers VN, Jezyk PF, Aguirre GD, Patterson DF (1983) Short-limbed dwarfism and ocular defects in the samoyed dog. J Am Vet Med Assoc 183:975–979
Miller PE, Galbreath EJ, Kehren JC, Steinberg H, Dubielzig RR (1998) Photoreceptor cell death by apoptosis in dogs with sudden acquired retinal degeneration syndrome. Am J Vet Res 59:149–152
Miyadera K, Kato K, Aguirre-Hernandez J, Tokuriki T, Morimoto K, Busse C, Barnett K, Holmes N, Ogawa H, Sasaki N, Mellersh CS, Sargan DR (2009) Phenotypic variation and genotype–phenotype discordance in canine cone–rod dystrophy with an RPGRIP1 mutation. Mol Vis 15:2287–2305
Miyadera K, Kato K, Boursnell M, Mellersh CS, Sargan DR (2012) Genome-wide association study in RPGRIP1−/− dogs identifies a modifier locus that determines the onset of retinal degeneration. Mamm Genome (under revision)
Mollet G, Salomon R, Gribouval O, Silbermann F, Bacq D, Landthaler G, Milford D, Nayir A, Rizzoni G, Antignac C, Saunier S (2002) The gene mutated in juvenile nephronophthisis type 4 encodes a novel protein that interacts with nephrocystin. Nat Genet 32:300–305
Moody JA, Famula TR, Sampson RC, Murphy KE (2005) Identification of microsatellite markers linked to progressive retinal atrophy in American Eskimo Dogs. Am J Vet Res 66:1900–1902
Mowat FM, Petersen-Jones SM, Williamson H, Williams DL, Luthert PJ, Ali RR, Bainbridge JW (2008) Topographical characterization of cone photoreceptors and the area centralis of the canine retina. Mol Vis 14:2518–2527
Narfström K, Wrigstad A, Nilsson SEG (1989) The Briard dog: a new animal model of congenital stationary night blindness. Br J Ophthalmol 73:750–756
Narfström K, Katz ML, Bragadottir R, Seeliger M, Boulanger A, Redmond TM, Caro L, Lai CM, Rakoczy PE (2003) Functional and structural recovery of the retina after gene therapy in the RPE65 null mutation dog. Invest Ophthalmol Vis Sci 44:1663–1672
Narfström K, Seeliger M, Lai CM, Vaegan, Katz M, Rakoczy EP, Reme C (2008) Morphological aspects related to long-term functional improvement of the retina in the 4 years following rAAV-mediated gene transfer in the RPE65 null mutation dog. Adv Exp Med Biol 613:139–146
Nathans J, Thomas D, Hogness DS (1986a) Molecular genetics of human color vision: the genes encoding blue, green, and red pigments. Science 232:193–202
Nathans J, Piantanida TP, Eddy RL, Shows TB, Hogness DS (1986b) Molecular genetics of inherited variation in human color vision. Science 232:203–210
Nawrot M, Liu T, Garwin GG, Crabb JW, Saari JC (2006) Scaffold proteins and the regeneration of visual pigments. Photochem Photobiol 82:1482–1488
Nelson D, MacMillan A (1983) Multifocal retinal dysplasia in field trial Labrador retrievers. J Am Anim Hosp Assoc 19:388
Nevet MJ, Shalev SA, Zlotogora J, Mazzawi N, Ben-Yosef T (2010) Identification of a prevalent founder mutation in an Israeli Muslim Arab village confirms the role of PRCD in the aetiology of retinitis pigmentosa in humans. J Med Genet 47:533–537
O’Toole D, Roberts S, Nunamaker C (1992) Sudden acquired retinal degeneration (‘silent retina syndrome’) in two dogs. Vet Rec 130:157–161
Olsson M, Meadows JR, Truve K, Rosengren Pielberg G, Puppo F, Mauceli E, Quilez J, Tonomura N, Zanna G, Docampo MJ, Bassols A, Avery AC, Karlsson EK, Thomas A, Kastner DL, Bongcam-Rudloff E, Webster MT, Sanchez A, Hedhammar A, Remmers EF, Andersson L, Ferrer L, Tintle L, Lindblad-Toh K (2011) A novel unstable duplication upstream of HAS2 predisposes to a breed-defining skin phenotype and a periodic fever syndrome in Chinese Shar-Pei dogs. PLoS Genet 7:e1001332
Otto E, Hoefele J, Ruf R, Mueller AM, Hiller KS, Wolf MT, Schuermann MJ, Becker A, Birkenhager R, Sudbrak R, Hennies HC, Nurnberg P, Hildebrandt F (2002) A gene mutated in nephronophthisis and retinitis pigmentosa encodes a novel protein, nephroretinin, conserved in evolution. Am J Hum Genet 71:1161–1167
Parker HG, Kim LV, Sutter NB, Carlson S, Lorentzen TD, Malek TB, Johnson GS, DeFrance HB, Ostrander EA, Kruglyak L (2004) Genetic structure of the purebred domestic dog. Science 304:1160–1164
Parker HG, Kukekova AV, Akey DT, Goldstein O, Kirkness EF, Baysac KC, Mosher DS, Aguirre GD, Acland GM, Ostrander EA (2007) Breed relationships facilitate fine-mapping studies: a 7.8-kb deletion cosegregates with Collie eye anomaly across multiple dog breeds. Genome Res 17:1562–1571
Parker HG, VonHoldt BM, Quignon P, Margulies EH, Shao S, Mosher DS, Spady TC, Elkahloun A, Cargill M, Jones PG, Maslen CL, Acland GM, Sutter NB, Kuroki K, Bustamante CD, Wayne RK, Ostrander EA (2009) An expressed fgf4 retrogene is associated with breed-defining chondrodysplasia in domestic dogs. Science 325:995–998
Parry HB (1953a) Degenerations of the dog retina. I. Structure and development of the retina of the normal dog. Br J Ophthalmol 37:385–404
Parry HB (1953b) Degenerations of the dog retina. II. Generalized progressive atrophy of hereditary origin. Br J Ophthalmol 37:487–502
Parry HB (1954a) Degenerations of the dog retina. IV. Retinopathies associated with dog distemper-complex virus infections. Br J Ophthalmol 38:295–309
Parry HB (1954b) Degenerations of the dog retina. VI. Central progressive atrophy with pigment epithelial dystrophy. Br J Ophthalmol 38:653–668
Parry HB, Tansley K, Thomson LC (1953) The electroretinogram of the dog. J Physiol 120:28–40
Parry HB, Tansley K, Thomson LC (1955) Electroretinogram during development of hereditary retinal degeneration in the dog. Br J Ophthalmol 39:349–352
Parry DA, Toomes C, Bida L, Danciger M, Towns KV, McKibbin M, Jacobson SG, Logan CV, Ali M, Bond J, Chance R, Swendeman S, Daniele LL, Springell K, Adams M, Johnson CA, Booth AP, Jafri H, Rashid Y, Banin E, Strom TM, Farber DB, Sharon D, Blobel CP, Pugh EN Jr, Pierce EA, Inglehearn CF (2009) Loss of the metalloprotease ADAM9 leads to cone–rod dystrophy in humans and retinal degeneration in mice. Am J Hum Genet 84:683–691
Parshall CJ, Wyman M, Nitroy S, Acland G, Aguirre G (1991) Photoreceptor dysplasia: an inherited progressive retinal atrophy of Miniature Schnauzer dogs. Progr Vet Comp Ophthalmol 1:187–203
Petersen-Jones SM, Entz DD, Sargan DR (1999) cGMP phosphodiesterase-α mutation causes progressive retinal atrophy in the cardigan Welsh corgi dog. Invest Ophthalmol Vis Sci 40:1637–1644
Priat C, Hitte C, Vignaux F, Renier C, Jiang Z, Jouquand S, Chéron A, André C, Galibert F (1998) A whole-genome radiation hybrid map of the dog genome. Genomics 54:361–378
Pugh EN Jr (1999) Variability in single photon responses: a cut in the Gordian knot of rod phototransduction? Neuron 23:205–208
Ray K, Baldwin VJ, Acland GM, Blanton SH, Aguirre GD (1994) Cosegregation of codon 807 mutation of the canine rod cGMP phosphodiesterase ß gene and rcd1. Invest Ophthalmol Vis Sci 35:4291–4299
Ray K, Acland GM, Aguirre GD (1996) Nonallelism of erd and prcd and exclusion of the canine RDS/peripherin gene as a candidate for both retinal degeneration loci. Invest Ophthalmol Vis Sci 37:783–794
Ray K, Wang W, Czarnecki J, Zhang Q, Acland GM, Aguirre GD (1999) Strategies for identification of mutations causing hereditary retinal diseases in dogs: evaluation of opsin as a candidate gene. J Hered 90:133–137
Roberts S (1969) The collie eye anomaly. J Am Vet Med Assoc 155:859–878
Ropstad EO, Bjerkas E, Narfstrom K (2007) Clinical findings in early onset cone–rod dystrophy in the standard wire-haired dachshund. Vet Ophthalmol 10:69–75
Ropstad EO, Narfstrom K, Lingaas F, Wiik C, Bruun A, Bjerkas E (2008) Functional and structural changes in the retina of wire-haired dachshunds with early-onset cone–rod dystrophy. Invest Ophthalmol Vis Sci 49:1106–1115
Rubin L (1968) Heredity of retinal dysplasia in bedlington terriers. J Am Vet Med Assoc 152:260–262
Rubin L, Bourns T, Lord L (1967) Hemeralopia in dogs: heredity of hemeralopia in Alaskan malamutes. Am J Vet Res 28:355–357
Runte M, Dekomien G, Epplen JT (2000) Evaluation of RDS/Peripherin and ROM1 as candidate genes in generalised progressive retinal atrophy and exclusion of digenic inheritance. Anim Genet 31:223–227
Salmon Hillbertz NH, Isaksson M, Karlsson EK, Hellmen E, Pielberg GR, Savolainen P, Wade CM, von Euler H, Gustafson U, Hedhammar A, Nilsson M, Lindblad-Toh K, Andersson L, Andersson G (2007) Duplication of FGF3, FGF4, FGF19 and ORAOV1 causes hair ridge and predisposition to dermoid sinus in Ridgeback dogs. Nat Genet 39:1318–1320
Sidjanin DJ, Lowe JK, McElwee JL, Milne BS, Phippen TM, Sargan DR, Aguirre GD, Acland GM, Ostrander EA (2002) Canine CNGB3 mutations establish cone degeneration as orthologous to the human achromatopsia locus ACHM3. Hum Mol Genet 11:1823–1833
Sidjanin DJ, Miller B, Kijas J, McElwee J, Pillardy J, Malek J, Pai G, Feldblyum T, Fraser C, Acland G, Aguirre G (2003) Radiation hybrid map, physical map, and low-pass genomic sequence of the canine prcd region on CFA9 and comparative mapping with the syntenic region on human chromosome 17. Genomics 81:138–148
Stone EM (2007) Leber congenital amaurosis—a model for efficient genetic testing of heterogeneous disorders: LXIV Edward Jackson Memorial Lecture. Am J Ophthalmol 144:791–811
Suber ML, Pittler SJ, Qin N, Wright GC, Holcombe V, Lee RH, Craft CM, Lolley RN, Baehr W, Hurwitz RL (1993) Irish setter dogs affected with rod/cone dysplasia contain a nonsense mutation in the rod cGMP phosphodiesterase ß-subunit gene. Proc Natl Acad Sci USA 90:3968–3972
Sutter NB, Eberle MA, Parker HG, Pullar BJ, Kirkness EF, Kruglyak L, Ostrander EA (2004) Extensive and breed-specific linkage disequilibrium in Canis familiaris. Genome Res 14:2388–2396
Turney C, Chong NH, Alexander RA, Hogg CR, Fleming L, Flack D, Barnett KC, Bird AC, Holder GE, Luthert PJ (2007) Pathological and electrophysiological features of a canine cone–rod dystrophy in the miniature longhaired dachshund. Invest Ophthalmol Vis Sci 48:4240–4249
Vainisi SJ, Schmidt GM, West CS et al (1983) Metabolic toxic retinopathy preliminary report. Trans Am Coll Vet Ophthalmol 14:76–81
van der Woerdt A, Nasisse M, Davidson M (1991) Sudden acquired retinal degeneration in the dog: clinical and laboratory in 36 cases. Prog Vet Comp Ophthalmol 1:11–18
van Wijk E, van der Zwaag B, Peters T, Zimmermann U, Te Brinke H, Kersten FF, Marker T, Aller E, Hoefsloot LH, Cremers CW, Cremers FP, Wolfrum U, Knipper M, Roepman R, Kremer H (2006) The DFNB31 gene product whirlin connects to the Usher protein network in the cochlea and retina by direct association with USH2A and VLGR1. Hum Mol Genet 15:751–765
van Wijk E, Kersten FF, Kartono A, Mans DA, Brandwijk K, Letteboer SJ, Peters TA, Marker T, Yan X, Cremers CW, Cremers FP, Wolfrum U, Roepman R, Kremer H (2009) Usher syndrome and Leber congenital amaurosis are molecularly linked via a novel isoform of the centrosomal ninein-like protein. Hum Mol Genet 18:51–64
Veske A, Nilsson SEG, Narfström K, Gal A (1999) Retinal dystrophy of Swedish briard/briard-beagle dogs is due to a 4-bp deletion in RPE65. Genomics 57:57–61
Vonholdt BM, Pollinger JP, Lohmueller KE, Han E, Parker HG, Quignon P, Degenhardt JD, Boyko AR, Earl DA, Auton A, Reynolds A, Bryc K, Brisbin A, Knowles JC, Mosher DS, Spady TC, Elkahloun A, Geffen E, Pilot M, Jedrzejewski W, Greco C, Randi E, Bannasch D, Wilton A, Shearman J, Musiani M, Cargill M, Jones PG, Qian Z, Huang W, Ding ZL, Zhang YP, Bustamante CD, Ostrander EA, Novembre J, Wayne RK (2010) Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 464:898–902
Wang W, Acland GM, Ray K, Aguirre GD (1999) Evaluation of cGMP-phosphodiesterase (PDE) subunits for causal association with rod–cone dysplasia 2 (rcd2), a canine model of abnormal retinal cGMP metabolism. Exp Eye Res 69:445–453
Wang P, Zangerl B, Werner P, Mauldin EA, Casal ML (2011) Familial cutaneous lupus erythematosus (CLE) in the German shorthaired pointer maps to CFA18, a canine orthologue to human CLE. Immunogenetics 63:197–207
Wiik AC, Wade C, Biagi T, Ropstad EO, Bjerkas E, Lindblad-Toh K, Lingaas F (2008) A deletion in nephronophthisis 4 (NPHP4) is associated with recessive cone–rod dystrophy in standard wire-haired dachshund. Genome Res 18(9):1415–1421
Wilbe M, Jokinen P, Truve K, Seppala EH, Karlsson EK, Biagi T, Hughes A, Bannasch D, Andersson G, Hansson-Hamlin H, Lohi H, Lindblad-Toh K (2010) Genome-wide association mapping identifies multiple loci for a canine SLE-related disease complex. Nat Genet 42:250–254
Wood SH, Ke X, Nuttall T, McEwan N, Ollier WE, Carter SD (2009) Genome-wide association analysis of canine atopic dermatitis and identification of disease related SNPs. Immunogenetics 61:765–772
Woodford B, Liu Y, Fletcher R, Chader G, Farber D, Santos-Anderson R, Tso M (1982) Cyclic nucleotide metabolism in inherited retinopathy in collies: a biochemical and histochemical study. Exp Eye Res 34:703–714
Wrigstad A (1994) Hereditary dystrophy of the retina and the retinal pigment epithelium in a strain of briard dogs: a clinical, morphological and electrophysiological study. In: Linköping University Medical Dissertations
Wrigstad A, Narfstrom K, Nilsson SEG (1994) Slowly progressive changes of the retina and retinal pigment epithelium in briard dogs with hereditary congenital night blindness and partial day blindness. a morphological study. Doc Ophthalmol 87:337–354
Zangerl B, Zhang Q, Pearce-Kelling SE, Aguirre GD (2002) Molecular cloning, characterization and mapping of the canine glucocorticoid receptor DNA binding factor 1 (GRLF1). GENE 294:167–176
Zangerl B, Goldstein O, Philp AR, Lindauer SJ, Pearce-Kelling SE, Mullins RF, Graphodatsky AS, Ripoll D, Felix JS, Stone EM, Acland GM, Aguirre GD (2006) Identical mutation in a novel retinal gene causes progressive rod–cone degeneration in dogs and retinitis pigmentosa in humans. Genomics 88:551–563
Zangerl B, Johnson JL, Pillardy J, Sun Q, Andre C, Galibert F, Acland GM, Aguirre GD (2009) Comparative genomic mapping of uncharacterized canine retinal ESTs to identify novel candidate genes for hereditary retinal disorders. Mol Vis 15:927–936
Zangerl B, Wickstrom K, Slavik J, Lindauer SJ, Ahonen S, Schelling C, Lohi H, Guziewicz KE, Aguirre GD (2010) Assessment of canine BEST1 variations identifies new mutations and establishes an independent bestrophinopathy model (cmr3). Mol Vis 16:2791–2804
Zeiss CJ, Acland GM, Aguirre GD (1999) Retinal pathology of canine X-linked progressive retinal atrophy, the locus homologue of RP3. Invest Ophthalmol Vis Sci 40:3292–3304
Zeng R, Farias FH, Johnson GS, McKay SD, Schnabel RD, Decker JE, Taylor JF, Mann CS, Katz ML, Johnson GC, Coates JR, O’Brien DP (2011) A truncated retrotransposon disrupts the GRM1 coding sequence in Coton de Tulear dogs with Bandera’s neonatal ataxia. J Vet Intern Med 25:267–272
Zernant J, Kulm M, Dharmaraj S, den Hollander AI, Perrault I, Preising MN, Lorenz B, Kaplan J, Cremers FP, Maumenee I, Koenekoop RK, Allikmets R (2005) Genotyping microarray (disease chip) for Leber congenital amaurosis: detection of modifier alleles. Invest Ophthalmol Vis Sci 46:3052–3059
Zhang Q, Acland GM, Parshall CJ, Haskell J, Ray K, Aguirre GD (1998) Characterization of canine photoreceptor phosducin cDNA and identification of a sequence variant in dogs with photoreceptor dysplasia. Gene 215:231–239
Zhang Q, Ray K, Acland GM, Czarnecki JM, Aguirre GD (2000) Molecular cloning, characterization and expression of a novel retinal clusterin-like protein cDNA. Gene 243:151–160
Zhang Q, Acland GM, Zangerl B, Johnson JL, Mao Z, Zeiss CJ, Ostrander EA, Aguirre GD (2001) Fine mapping of canine XLPRA establishes homology of the human and canine RP3 intervals. Invest Ophthalmol Vis Sci 42:2466–2471
Zhang Q, Acland GM, Wu WX, Johnson JL, Pearce-Kelling S, Tulloch B, Vervoort R, Wright AF, Aguirre GD (2002) Different RPGR exon ORF15 mutations in Canids provide insights into photoreceptor cell degeneration. Hum Mol Genet 11:993–1003
Zhou Z, Sheng X, Zhang Z, Zhao K, Zhu L, Guo G, Friedenberg SG, Hunter LS, Vandenberg-Foels WS, Hornbuckle WE, Krotscheck U, Corey E, Moise NS, Dykes NL, Li J, Xu S, Du L, Wang Y, Sandler J, Acland GM, Lust G, Todhunter RJ (2010) Differential genetic regulation of canine hip dysplasia and osteoarthritis. PLoS One 5:e13219
Zhu L, Jang GF, Jastrzebska B, Filipek S, Pearce-Kelling SE, Aguirre GD, Stenkamp RE, Acland GM, Palczewski K (2004) A naturally occurring mutation of the opsin gene (T4R) in dogs affects glycosylation and stability of the G protein-coupled receptor. J Biol Chem 279:53828–53839
Acknowledgments
KM acknowledges The Kennel Club Charitable Trust (RG55218) for research funds; Fitzwilliam College, University of Cambridge, The University of Tokyo, British Council Japan Association for scholarships. Much of KM’s work is the result of the exceptional mentorship provided by Drs. David Sargan at the University of Cambridge, Cathryn Mellersh at the Animal Health Trust, Nobuo Sasaki and Kumiko Kato at The University of Tokyo, and Hiroyuki Ogawa at the Japan Animal Referral Medical Center. GMA and GDA acknowledge NEI/NIH grants EY-01244, EY06855, EY13132, and EY13729, The Foundation Fighting Blindness, The ONCE International Prize for Research & Development in Biomedicine and New Technologies for the Blind, and The Van Sloun Fund for Canine Genetic Research for their generous support. They are indebted to Sue Pearce-Kelling for her many contributions in all aspects of the research, to our scientific colleagues—research scientists, postdoctoral fellows, graduate students, and research technicians, and the staff of the RDSF—and to the dog owners for their participation in research. The authors thank Optigen LLC for disclosing unpublished data.
Disclosure
GMA and GDA are founding members and owners of OptiGen LLC, a company that carries out DNA testing for a large number of inherited eye diseases in dogs, and hold patents for some of these tests.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Miyadera, K., Acland, G.M. & Aguirre, G.D. Genetic and phenotypic variations of inherited retinal diseases in dogs: the power of within- and across-breed studies. Mamm Genome 23, 40–61 (2012). https://doi.org/10.1007/s00335-011-9361-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00335-011-9361-3