
Abstract
Objectives
The high variability of hypertrophic cardiomyopathy (HCM) genetic phenotypes has prompted the establishment of risk-stratification systems that predict the risk of a positive genetic mutation based on clinical and echocardiographic profiles. This study aims to improve mutation-risk prediction by extracting cardiovascular magnetic resonance (CMR) morphological features using a deep learning algorithm.
Methods
We recruited 198 HCM patients (48% men, aged 47 ± 13 years) and divided them into training (147 cases) and test (51 cases) sets based on different genetic testing institutions and CMR scan dates (2012, 2013, respectively). All patients underwent CMR examinations, HCM genetic testing, and an assessment of established genotype scores (Mayo Clinic score I, Mayo Clinic score II, and Toronto score). A deep learning (DL) model was developed to classify the HCM genotypes, based on a nonenhanced four-chamber view of cine images.
Results
The areas under the curve (AUCs) for the test set were Mayo Clinic score I (AUC: 0.64, sensitivity: 64.29%, specificity: 47.83%), Mayo Clinic score II (AUC: 0.70, sensitivity: 64.29%, specificity: 65.22%), Toronto score (AUC: 0.74, sensitivity: 75.00%, specificity: 56.52%), and DL model (AUC: 0.80, sensitivity: 85.71%, specificity: 69.57%). The combination of the DL and the Toronto score resulted in a significantly higher predictive performance (AUC = 0.84, sensitivity: 83.33%, specificity: 78.26%), compared with Mayo I (p = 006), Mayo II (p = 022), and Toronto score (p = 0.029).
Conclusions
The combination of the DL model, based on nonenhanced cine CMR images and the Toronto score yielded significantly higher diagnostic performance in detecting HCM mutations.
Key Points
• Deep learning method could enable the extraction of image features from cine images.
• Deep learning method based on cine images performed better than established scores in identifying HCM patients with positive genotypes.
• The combination of the deep learning method based on cine images and the Toronto score could further improve the performance of the identification of HCM patients with positive genotypes.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Hypertrophic cardiomyopathy (HCM) is the most frequent cause of sudden cardiac death (SCD) in young adults [1]. HCM is characterized by heterogeneities in morphological expressions and clinical courses [2]. Additionally, it is the most common autosomal dominant inherited cardiovascular disorder. In up to 60% of HCM patients, more than 1400 mutations in genes encoding sarcomere proteins have been detected [3]. Genetic testing has a limited impact on the treatment strategies for individual patients, but a positive genetic test result could confirm the etiology of the disease and enable cascade genetic screening of their relatives [4]. Moreover, compared with conventional regular clinical screening, the addition of genetic testing is cost-effective [5]. However, based on the current genetic testing studies in HCM populations, the yield of causative mutations was viable (15–70%) [6,7,8]. Thus, it should be noted that the selection of patients who have high probability of positive HCM genotypes can maximize the cost-effectiveness for genetic testing.
Although precise correlations have not been established between the phenotype and genotype in HCM patients, patients with mutations were considered to show significant differences in both clinical and imaging features compared to those without mutations, such as a family history of HCM and SCD, and the left ventricular maximal wall thickness (LVMWT) [9, 10]. Previous studies [11] have proposed several scores to predict positive genotypes in HCM based on the linear regression modeling of clinical and imaging variables; however, these scores were not fully validated in clinical practice and could not directly reflect the dynamic and physiological complexity of the myocardium.
Radiomics have demonstrated the potential to achieve or surpass visual detection in image analysis. Cardiovascular magnetic resonance (CMR) is not only applicable to the clinical diagnosis and management of HCM [4] owing to its excellent high spatial resolution and superior contrast ability, it also lends itself to radiomic analysis. Radiomic signatures based on magnetic resonance sequences have been associated with particular genetic expressions in glioma, EGFR expression [12], and p53 status [13].
Deep learning (DL) is a subset of machine learning algorithms that can learn features and transform them into class labels for classification. DL is currently reported to recognize risk stratification in a variety of CMR studies [14,15,16,17]. In particular, nonenhanced cardiac cine sequence is routinely used in clinical practice to visualize cardiac motion, which is also free from renal impairment due to contrast agents. DL based on nonenhanced cardiac cine image–based DL has accurate fully automated LV segmentations [18] and can efficiently detect myocardial infarction [14]. In this study, we aim to improve the HCM mutation-risk prediction by developing a nonenhanced cine image–based DL model to enable cost-effective genetic testing for HCM patients.
Material and methods
Study population
The study population consisted of 198 HCM patients from January 2012 to December 2013 in our referral center. HCM was defined as the presence of increased LV wall thickness (≥ 15 mm or ≥ 13 mm with a family history) that is not solely explained by abnormal loading conditions. All patients underwent CMR examinations and evaluations of genotype status by three established scores (Mayo Clinic score I, Mayo Clinic score II, Toronto score) [11, 19, 20] to predict HCM genotype (Table 1). Patients with HCM phenocopies (n = 4) and poor image quality (n = 6) were excluded. All patients provided informed consent, and the local ethics committee approved this retrospective observational study (2017-923).
Genetic testing
Genetic testing was performed at two centers: Biotechnology Corporation (n = 147) and the State Key Laboratory of Cardiovascular Disease in Fuwai Hospital (n = 51). Peripheral blood–derived DNA from all HCM patients were used for panel sequencing of the eight sarcomere protein-encoding genes including myosin-binding protein C (MYBPC3), β-myosin heavy chain (MYH7), essential and regulatory myosin light chains (MYL2, MYL3), cardiac troponin T (TNNT2), cardiac troponin I (TNNI3), α-tropomyosin (TPM1), and cardiac actin (ACTC1), as well as 3 HCM phenocopy genes (GLA for Fabry disease, LAMP2 for Danon disease, and PRKAG2 for PRKAG2 Cardiomyopathy). All coding exons and their flanking intronic regions were captured using Agilent probes (Agilent Technologies) and analyzed using the Illumina HiSeq X-ten platform (n = 147) and HiSeq 2500 (n = 51) (Illumina Inc.), respectively. Data analysis was performed using a custom bioinformatics workflow [21] (details are provided in Supplement 1).
CMR cine data
CMR images of all the HCM patients were obtained using a clinical 1.5-T MR scanner (MAGNETOM Avanto, Siemens Healthcare), which implemented an electrocardiographic and respiratory gating with a unified protocol. A balanced steady-state free precession cine sequence with a breath-hold technique was performed in the LV two-chamber, three-chamber, four-chamber, and short-axis orientation. The typical imaging parameters were repetition time (TR) = 2.8–3.0 ms, echo time (TE) = 1.1–1.5 ms, flip angle = 60–70°, temporal resolution = 30–55 ms, field of view (FOV) = 360 × 315 mm2, matrix = 192 × 162, slice thickness = 8 mm, and slice gap = 2 mm. The LVMWT and left ventricular posterior wall thickness (LVPWT) were measured at the thickest segment on the short-axis cine image in the LV end-diastole and the ratio of LVWMT/LVPWT was calculated. The septal morphological subtypes (sigmoid, reverse curve, apical, and neutral) were assessed from the three-chamber long-axis views [22].
Cine image segmentation
Cine images of a four-chamber view were retrieved from the picture archiving and communication system and loaded onto the free ITK-SNAP 3.6.0 software. The end-systolic and end-diastolic phases of the four-chamber-view cine images in the DICOM format were selected for further segmentation by a radiologist (G.Y. with 10 years of experience in CMR). Regions of interest (ROIs) were manually drawn, encircling the LV myocardium on end-systolic and end-diastolic phases. Furthermore, the propagation of the ROIs to the entire cardiac cycle was completed using the DL model, as detailed in the next subsection.
Deep learning model
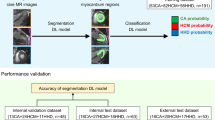
In this study, the establishment of the DL network was divided into three stages, as shown in Fig. 1. The original cine images were used as the input to the network, and the probability of a gene-positive expression was obtained as the output. In the first stage, the ROIs of the end-systolic and end-diastolic LV myocardium were propagated to all phases throughout the cardiac cycle using a DL image segmentation model DeeplabV3+ [23]. In the second stage, to form a three-channel input image, the original cine image (occupying two channels) and segmentation (occupying one channel) were combined to obtain both LV myocardial texture and morphological information. The three-channel input image was then input into the frozen-weight InceptionResnetV2 [24] model, which was pre-trained on the ImageNet dataset [25] to extract image features (n = 1536) in the ROI regions (Fig. 2). Moreover, to minimize excessive redundant information, these features underwent supervised dimensionality reduction (from 1536 to 32) through the last two fully connected layers in this model. In the third stage, predictions were performed by analyzing the image features obtained from the entire cardiac cycle using a long short-term memory (LSTM) network. The image features of 25 cine images (25*32) were respectively loaded onto the LSTM network [26]; the network was able to learn the subtle changes between different frames of a time-dependent series so that a regression analysis could determine the HCM mutation probability. Figure 3 presents the visualization of DL model from two representative cases.

Full process of the deep learning network. Firstly, the original images with ROI encircling the LV myocardium on end-diastolic and end-systolic phases were propagated to the entire cardiac cycle by using the deeplabV3 + model. Secondly, the ROIs were cropped and combined with the original cine images; image features were subsequently extracted using a classification model: InceptionResnetV2 model. Finally, the image features of the entire frames were placed into a LSTM model to evaluate the motion state of each patient and to obtain the probability of an HCM gene-positive expression. Abbreviations: HCM, hypertrophic cardiomyopathy; LV, left ventricular; ROIs, regions of interest; LSTM, long short-term memory

Flow chart for image feature extraction. In this part, single image features were extracted from the pre-trained InceptionResnetV2 model and underwent targeted dimensionality reduction (from 1536 to 32) by the last two fully connected layers

Four-chamber view of cine images (odd frames) and visualization of model features from a genotype-positive (upper two rows) and a genotype-negative (lower two rows) HCM patient, respectively. The color differences represented the intensity of each pixel in the entire model, which may reflect the probability of HCM gene expression in each patient. Red indicated a higher probability (p = 0.87) and blue indicated a lower probability (p = 0.28). Abbreviations: HCM, hypertrophic cardiomyopathy
The data set was divided into a training set (n = 147) and a test set (n = 51) based on different genetic testing institutions and CMR scan dates (2012 and 2013, respectively) (Table 2). To evaluate the stability of the DL model, we repeated the experiment using random 10-fold cross-validation for the training set. All three models in the network were trained by an Adam optimizer (learning rate = 0.0001) [27], using cross-entropy as a loss function. A total of twenty epochs were trained in each model, and the results were retained to minimize the loss of the training set. The DL network was implemented using the Keras [28] DL toolkit in Python, version 3.6. Moreover, model training was performed on graphical processor units (TITAN XP, NVIDIA).
Statistical analysis
Data were presented as a mean ± standard deviation, median (quartiles 25–75%), or n (%), as appropriate. The Kolmogorov–Smirnov test was used to assess the normal distribution of continuous data. The Student t test was utilized to compare the continuous variables between the two groups. Nonparametric parameters were compared using the Mann–Whitney U test. Frequencies were compared using the chi-squared test or Fisher’s exact test, as appropriate. The predictive performance of the Mayo Clinic score I, Mayo Clinic score II, Toronto scores, and DL model was evaluated through receiver-operating characteristic (ROC) analysis to identify positive HCM genotypes. Logistic regression was performed using the scikit-learn package in Python. All statistical calculations were performed using the R software version 3.4.
Results
Clinical and CMR parameters
The study population consisted of 198 HCM patients (n = 198, 48% men; aged 47 ± 13 years). Unlike genotype-negative patients, genotype-positive patients included a significantly higher proportion of female patients (37.76% vs 24.00%, p = 0.036), patients aged < 45 years at the time of diagnosis (48.98% vs 35.00%, p = 0.046), and patients with a family history of HCM (29.59% vs 15.00%, p = 0.014). Moreover, these patients had a significantly lower prevalence of hypertension (23.47% vs 47.00%, p = 0.001) than genotype-negative patients. In terms of CMR data, the HCM morphological subtypes were categorized as follows: sigmoid (n = 111), reverse curve (n = 56), apical (n = 27), and neutral (n = 4). Unlike genotype-negative patients, genotype-positive patients exhibited greater LVMWT (23.70 ± 5.66 mm vs 21.53 ± 5.56 mm, p = 0.007) and higher LVMWT/LVPWT ratios (2.90 ± 1.02 vs 2.38 ± 0.68, p < 0.001) (Table 3).
HCM genotype
The overall yield of the genetic testing in this study was 49.49% (98/198). In this subset of genotype-positive patients, variants were distributed in MYBPC3 (n = 41; 41.84%), MYH7 (n = 34; 34.69%), MYL2 (n = 2; 2.04%), TPM1 (n = 2; 2.04%), TNNI3 (n = 5; 5.10%), TNNT2 (n = 3; 3.06%), ACTC1 (n = 3; 3.06%), MYL3 (n = 3; 3.06%), and compound mutations (n = 5; 5.10%).(Table 4 and Supplement 2).
Model performance
Training set
To evaluate the stability of the models, we used the network to perform a 10-fold cross-validation on the internal dataset. The area under the curve (AUC) value for the Mayo I score and the Mayo II score, the Toronto score, and DL model were 0.63 ± 0.04, 0.67 ± 0.04, 0.68 ± 0.04, and 0.81 ± 0.01, respectively.
Test set
The diagnostic performance in the test set was computed at the optimal thresholds that maximize the Youden index, which were as follows (Table 5 and Fig. 4): Mayo Clinic score I (AUC: 0.64, sensitivity: 64.29%, specificity: 47.83%,, accuracy: 56.86%), Mayo Clinic score II (AUC: 0.70, sensitivity: 64.29%, specificity: 65.22%, accuracy: 64.71%), Toronto score (AUC: 0.74, sensitivity: 75.00%, specificity: 56.52%, false-positive rate: 43.48%, accuracy: 66.67%), and DL model (AUC: 0.80, sensitivity: 85.71%, specificity: 69.57%, accuracy: 78.43%). Although the DL model exhibited a higher predictive performance, no statistical significance was achieved compared with the Mayo Clinic score I, Mayo Clinic score II, and Toronto score. However, the combination of DL and Toronto score resulted in significantly higher predictive performance (AUC = 0.84, sensitivity: 83.33%, specificity: 78.26%, accuracy: 84.31%) compared with Mayo I (p = 006), Mayo II (p = 022), and Toronto score (p = 0.029). Furthermore, the false-positive rate was 52.17%, 34.78%, 43.48%, and 30.43% in Mayo I, Mayo II, Toronto score, and DL model, respectively, resulting in 12, 8, 10, and 7 genotype-negative HCM patients misclassified in the genotype-positive group, respectively. The combination of the DL model and Toronto score resulted in only five genotype-negative HCM patients misclassified in the genotype-positive group.

ROC curves of the three established scores, DL, and DL + Toronto score for the reflection of HCM genotypes in the test set. Abbreviations: AUC, area under the (receiver-operating characteristic) curve; DL, deep learning; HCM, hypertrophic cardiomyopathy; ROC, receiver-operating characteristic; T, Toronto score
Discussion
In this study, to maximize the cost-effectiveness of HCM genetic testing and explore the potential value of CMR in reflecting HCM genotype status, we developed a nonenhanced cine CMR image–based DL model. The underlying hypothesis is that cine images can provide internal myocardial structural and motional information, which is routinely used in clinical practice, without the administration of contrast agents. Our results indicate that a HCM mutation may be predicted with an AUC of 0.80 and an accuracy of 78.43% using the DL model. Reasonable consistency of the results was noted among the 10-fold cross-validation, thereby suggesting a stable network performance. In addition, the combination of the DL model and the Toronto score (with an AUC of 0.84 and an accuracy of 84.31%) yielded significantly higher diagnostic performance than that of a single score.
Clinical and CMR parameters
The overall yield of genetic testing was 49.5%, which was within the range of yields reported in previous literature (15–70%) [6,7,8]. Similar to the results of previous studies [29], 76.5% of causative mutations were detected in MYBPC3 and MYH7 in this study. Furthermore, several clinical and imaging parameters were reported to be associated with HCM genotypes. In this study, HCM patients with positive gene expressions were associated with diagnosis at a young age, high prevalence of family history of HCM, low prevalence of hypertension, and great LVMWT, which is consistent with previous literatures [9]. Three scores have been established to predict HCM genotypes by combining different clinical and imaging parameters. Mayo Clinic score I only considered age at diagnosis, LVMWT, and family history of HCM. Mayo Clinic score II added septal morphology, family history of SCD, and hypertension. In the Toronto score, further details, with different risk weights, were attached to independent predictor variables. Among the three traditional scores, the Toronto score provided the best predictive performance. However, the traditional scores had a limited accuracy (56.86–66.67%). This may be because the traditional scores were established based on the linear regression modeling of clinical and imaging variables and a single score cannot reflect the dynamic physiological complexity of this heart disease.
Deep learning model
Compared with a human visual inspection, the DL technique may detect subtle motional changes in cardiovascular diseases, e.g., myocardial infarction, with higher precision and sensitivity [14, 30]. The DL model was also used to achieve a fully automated and accurate LV functional analysis of CMR cine images [31]. In our DL model, the differences in the internal myocardial structure were reflected in the intensity changes in the myocardial features in each frame of the image. A.H. Ellims et al [32] detected more regional, but less diffuse myocardial fibrosis in HCM patients with genetic mutations than those without genetic mutations, suggesting different pathological features in HCM patients with positive and negative genotypes. Furthermore, the difference in myocardial motion was reflected in the temporal state of the myocardial features. This may be owing to the different HCM genetic status, which may lead to subtle differences in both the internal structure and motion states of the myocardium. It was emphasized that the combination of the DL model and Toronto score resulted in the best predictive performance with an AUC of 0.84 and a lower false-positive rate (approximately 20%) than single scores. This implies a significant complementation between internal myocardial characterization and clinical features.
Clinical implications
CMR should be considered for patients that fulfill the diagnostic criteria for HCM, to assess cardiac anatomy, ventricular function, and the presence and extent of myocardial fibrosis (IIa) [4]. However, CMR is underutilized in HCM, primarily owing to the length of the CMR examination, lack of clinicians experienced in cardiac imaging, and relatively high cost (approximately 286USD, 70% covered by social medical insurance in China). Thus, it is impractical to perform CMR for all HCM patients. We developed a DL model based on cine images of those patients who have undergone CMR examinations, to identify genotype information and assist in selecting patients with a higher probability of a positive genotype to maximize the cost-effectiveness of genetic testing. For example, using the combined DL model and Toronto score, five of fifty-one (< 10%) patients were misclassified in the genotype-positive group, which was significantly lower than the misclassifications by established scores (15–25%). The identification of causative mutations in an HCM probands could facilitate the detection of asymptomatic HCM patients and mutation carriers among their family members. Although the genotype-phenotype relationship in HCM has not been clearly established, it has been widely accepted that HCM patients with a positive genotype are associated with malignant prognosis, compared to those with a negative genotype [10, 33, 34]. Thus, the DL-based technique on nonenhanced CMR cine images also has the potential to improve the interpretation of the genotype-phenotype correlation in the HCM population.
Study limitation
We acknowledge that the preliminary study has several limitations. First, the DL network exhibits difficulty in obtaining stable image features directly from the data, owing to the limited dataset. A larger set of training and test data may facilitate further improvement. Second, multi-center validation was not performed because the available data were obtained from a single tertiary hospital. Third, we focused on the classification of positive and negative genotypes; therefore, the performance of LV segmentation is not evaluated in this study. An anatomically constrained neuronal network may improve the segmentation and performance of the DL model in future studies. Fourth, although CMR is characterized by multi-sequence imaging, only four-chamber-view cine images were analyzed in this study owing to their reliable position, and no radiation as well as renal fibrosis due to contrast agents. Other sequences in different orientations may be investigated in future studies.
Conclusion
The combination of the DL model, based on cine images and the Toronto score, may aid in identifying HCM patients with positive genotypes, which can also potentially enhance the interpretation of the genotype and phenotype on CMR in the HCM population. Multivendor, multi-center participation, and a larger sample size are imperative to evaluate the feasibility and clinical application of this model.
Abbreviations
- AUC:
-
Area under the (receiver-operating characteristic) curve
- CMR:
-
Cardiac magnetic resonance
- DL:
-
Deep learning
- FHHCM:
-
Family history of hypertrophic cardiomyopathy
- FHSCD:
-
Family history of sudden cardiac death
- HCM:
-
Hypertrophic cardiomyopathy
- LSTM:
-
Long short-term memory
- LVMWT:
-
Left ventricular maximal wall thickness
- LVPWT:
-
Left ventricular posterior wall thickness
- ROC:
-
Receiver-operating characteristic
- ROIs:
-
Regions of interest
- SCD:
-
sudden cardiac death
- VUS:
-
Variants of uncertain significance
References
Maron BJ (2002) Hypertrophic cardiomyopathy: a systematic review. JAMA 287:1308–1320
Maron BJ, Maron MS (2013) Hypertrophic cardiomyopathy. Lancet 381:242–255
Maron BJ, Maron MS, Semsarian C (2012) Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 60:705–715
Elliott PM, Anastasakis A, Borger MA et al (2014) 2014 ESC guidelines on diagnosis and management of hypertrophic cardiomyopathy: the task force for the diagnosis and management of hypertrophic cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J 35:2733–2779
Wordsworth S, Leal J, Blair E et al (2010) DNA testing for hypertrophic cardiomyopathy: a cost-effectiveness model. Eur Heart J 31:926–935
Van Driest SL, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ (2005) Sarcomeric genotyping in hypertrophic cardiomyopathy. Mayo Clin Proc 80:463–469
Andersen PS, Havndrup O, Hougs L et al (2009) Diagnostic yield, interpretation, and clinical utility of mutation screening of sarcomere encoding genes in Danish hypertrophic cardiomyopathy patients and relatives. Hum Mutat 30:363–370
Girolami F, Olivotto I, Passerini I et al (2006) A molecular screening strategy based on beta-myosin heavy chain, cardiac myosin binding protein C and troponin T genes in Italian patients with hypertrophic cardiomyopathy. J Cardiovasc Med (Hagerstown) 7:601–607
Lopes LR, Rahman MS, Elliott PM (2013) A systematic review and meta-analysis of genotype-phenotype associations in patients with hypertrophic cardiomyopathy caused by sarcomeric protein mutations. Heart 99:1800–1811
Lopes LR, Syrris P, Guttmann OP et al (2015) Novel genotype-phenotype associations demonstrated by high-throughput sequencing in patients with hypertrophic cardiomyopathy. Heart 101:294–301
Gruner C, Ivanov J, Care M et al (2013) Toronto hypertrophic cardiomyopathy genotype score for prediction of a positive genotype in hypertrophic cardiomyopathy. Circ Cardiovasc Genet 6:19–26
Li Y, Qian Z, Xu K et al (2018) MRI features predict p53 status in lower-grade gliomas via a machine-learning approach. Neuroimage Clin 17:306–311
Li Y, Liu X, Xu K et al (2018) MRI features can predict EGFR expression in lower grade gliomas: a voxel-based radiomic analysis. Eur Radiol 28:356–362
Zhang N, Yang G (2019) Deep learning for diagnosis of chronic myocardial infarction on nonenhanced cardiac cine MRI. Radiology 291:606–617
Baessler B, Luecke C, Lurz J et al (2018) Cardiac MRI texture analysis of T1 and T2 maps in patients with infarctlike acute myocarditis. Radiology 289:357–365
Cheng S, Fang M, Cui C et al (2018) LGE-CMR-derived texture features reflect poor prognosis in hypertrophic cardiomyopathy patients with systolic dysfunction: preliminary results. Eur Radiol 28:4615–4624
Bello GA, Dawes TJW, Duan J et al (2019) Deep learning cardiac motion analysis for human survival prediction. Nat Mach Intell 1:95–104
Bai W, Sinclair M, Tarroni G et al (2018) Automated cardiovascular magnetic resonance image analysis with fully convolutional networks. J Cardiovasc Magn Reson 20:65
Bos JM, Will ML, Gersh BJ, Kruisselbrink TM, Ommen SR, Ackerman MJ (2014) Characterization of a phenotype-based genetic test prediction score for unrelated patients with hypertrophic cardiomyopathy. Mayo Clin Proc 89:727–737
Van Driest SL, Ommen SR, Tajik AJ, Gersh BJ, Ackerman MJ (2005) Yield of genetic testing in hypertrophic cardiomyopathy. Mayo Clin Proc 80:739–744
Richards S, Aziz N, Bale S et al (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 17:405–424
Binder J, Ommen SR, Gersh BJ et al (2006) Echocardiography-guided genetic testing in hypertrophic cardiomyopathy: septal morphological features predict the presence of myofilament mutations. Mayo Clin Proc 81:459–467
Chen LC, Zhu Y, Papandreou G, Schroff F, Adam H (2018) Encoder-decoder with atrous separable convolution for semantic image segmentation. European Conference on Computer Vision (ECCV), Munich. Available via https://doi.org/10.1007/978-3-030-01234-2_49. Accessed 06 October 2018
Szegedy C, Ioffe S, Vanhoucke VAlemi A (2016) Inception-v4, Inception-ResNet and the impact of residual connections on learning. National Conference on Artificial Intelligence(AAAI) , Phoenix. Available via arXiv:1602.07261. Accessed: February 2016
Deng J, Dong W, Socher R, Li LJ, Li K, Li FF (2009) ImageNet: a large-scale hierarchical image database. Computer Vision & Pattern Recognition (CVPR), Miami. Available via https://doi.org/10.1109/CVPR.2009.5206848. Accessed 18 August 2009
Gers FA, Schmidhuber J, Cummins F (2000) Learning to forget: continual prediction with LSTM. Neural Comput 12:2451–2471
Kingma DP, Ba J (2015) Adam: a method for stochastic optimization. International Conference on Learning Representations (ICLR), San Diego. Available via arXiv:1412.6980. Accessed 2015
Chollet F (2018) Keras: the python deep learning library. Astrophysics Source Code Library. Available via https://keras.io/getting-started/faq/#how-should-i-cite-keras. Accessed June 2018
Bos JM, Towbin JA, Ackerman MJ (2009) Diagnostic, prognostic, and therapeutic implications of genetic testing for hypertrophic cardiomyopathy. J Am Coll Cardiol 54:201–211
Baessler B, Mannil M, Oebel S, Maintz D, Alkadhi H, Manka R (2018) Subacute and chronic left ventricular myocardial scar: accuracy of texture analysis on nonenhanced cine MR images. Radiology 286:103–112
Tao Q, Yan W, Wang Y et al (2019) Deep learning-based method for fully automatic quantification of left ventricle function from cine MR images: a multivendor, multicenter study. Radiology 290:81–88
Ellims AH, Iles LM, Ling LH et al (2014) A comprehensive evaluation of myocardial fibrosis in hypertrophic cardiomyopathy with cardiac magnetic resonance imaging: linking genotype with fibrotic phenotype. Eur Heart J Cardiovasc Imaging 15:1108–1116
Fujita T, Fujino N, Anan R et al (2013) Sarcomere gene mutations are associated with increased cardiovascular events in left ventricular hypertrophy: results from multicenter registration in Japan. JACC Heart Fail 1:459–466
Olivotto I, Girolami F, Ackerman MJ et al (2008) Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc 83:630–638
Funding
This paper is supported by the National Natural Science Foundation of China under Grant Nos. 81922040, 81930053, 81227901, 81527805, and 81772012; the major international (regional) joint research project of National Science Foundation of China under Grant No. 81620108015; Beijing Natural Science Foundation (under Grant No. 7182109); National Key Research and Development Plan of China (under Grant Nos. 2017YFA0205200, 2016YFA0100900, and 2016YFA0100902); and Youth Innovation Promotion Association CAS (Grant No. 2019136).
Author information
Authors and Affiliations
Contributions
Lu Li made contributions to the conception and design of the study; Lu Li and Hongyu Zhou drafted the manuscript; Kankan Zhao and Lu Li were responsible for statistical analysis of the data, Zhenyu Liu, Min-jie Lu, and Xiuyu Chen made critical revisions to the manuscript; Lu Li and Lei Song collected conventional CMR and genetic data; Gang Yin was in charge of image segmentation; Shihua zhao, Jie Tian, and Hairong Zheng made a contribution to study conduction. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Guarantor
The scientific guarantor of this publication is Shihua Zhao.
Conflict of interest
The authors of this manuscript declare no relationships with any companies whose products or services may be related to the subject matter of the article.
Statistics and biometry
No complex statistical methods were necessary for this paper.
Informed consent
Written informed consent was obtained from all subjects (patients) in this study.
Ethical approval
Institutional Review Board approval was obtained.
Methodology
• Retrospective
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
ESM 1
(DOCX 21.3 kb)
Rights and permissions
About this article

Cite this article
Zhou, H., Li, L., Liu, Z. et al. Deep learning algorithm to improve hypertrophic cardiomyopathy mutation prediction using cardiac cine images. Eur Radiol 31, 3931–3940 (2021). https://doi.org/10.1007/s00330-020-07454-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00330-020-07454-9