
Abstract
Key message
A large, 53-kbp, intact DNA fragment was inserted into the wheat ( Triticum aestivum L.) genome. FISH analyses of individual transgenic events revealed multiple insertions of intact fragments.
Abstract
Transferring large intact DNA fragments containing clusters of resistance genes or complete metabolic pathways into the wheat genome remains a challenge. In a previous work, we showed that the use of dephosphorylated cassettes for wheat transformation enabled the production of simple integration patterns. Here, we used the same technology to produce a cassette containing a 44-kb Arabidopsis thaliana BAC, flanked by one selection gene and one reporter gene. This 53-kb linear cassette was integrated in the bread wheat (Triticum aestivum L.) genome by biolistic transformation. Our results showed that transgenic plants harboring the entire cassette were generated. The inheritability of the cassette was demonstrated in the T1 and T2 generation. Surprisingly, FISH analysis performed on T1 progeny of independent events identified double genomic insertions of intact fragments in non-homoeologous positions. Inheritability of these double insertions was demonstrated by FISH analysis of the T1 generation. Relative conclusions that can be drawn from molecular or FISH analysis are discussed along with future prospects of the engineering of large fragments for wheat transformation or genome editing.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Wheat, rice and maize are the main crops that provide food for humans and feed for animals. Alone, wheat provides 20% of the calories consumed by humans (http://faostat.fao.org/site/339/default.aspx). Until recently, one of the breeders’ goals was to increase productivity to get past the predicted population peak in 2050. Recently published results (Gerland et al. 2014) suggest that the world population is unlikely to stop growing this century and can be expected to increase to 10 billion in 2100. Therefore, yield improvement appears to be a major goal for agriculture for the next 20 years. Even if classical breeding is still the most widely used method for wheat improvement worldwide, biotechnologies can provide research teams with functional validation tools for rapid understanding of gene functions, and breeders with new tools to achieve their selection objectives more efficiently. Although wheat was the last of the major cereals to be transformed, first by biolistics (Vasil et al. 1992) and some years later by Agrobacterium-mediated transformation (Cheng et al. 1997), advances in tissue culture, combined with improvements in transformation technology, have resulted in increased transformation efficiency, and today, the routine introduction of a transgene in this crop is feasible (for reviews see Sparks and Jones 2014; Spark et al. 2014). The efficiency of wheat transformation has been improved over the past 20 years with the goal of transferring a combination of one or two genes to the selective marker gene, but certain research programs require the transfer of large fragments, for example gene clusters for disease resistance, genes with particularly long 5′ regulatory sequences (Saidi et al. 2007; Hu et al. 2008) or for the importing of entire metabolic pathways that can require up to nine genes (Naqvi et al. 2010). To conserve the entire functionality of the transferred DNA, care must be taken to transfer a linear, unbroken large fragment with no rearrangements.
The transformation of plants using large DNA fragments was achieved using binary bacterial artificial chromosome (BIBAC) technology, biolistic transformation or bioactive beads transformation technology. Hamilton et al. (1996) described the introduction of 30 and 150 kb of yeast and human genomic DNA, respectively, into the tobacco genome using a binary-BAC system strategy. Using this strategy, five tobacco independent events were obtained, which appeared to contain the entire intact 150-kb human DNA fragment. Additional results in tomato showed that transgenic events obtained using the same BIBAC strategy (Hamilton 1997) were stable through several generations without gene silencing (Frary and Hamilton 2001). In the same way, transformation-competent artificial chromosomes (TAC) that can accept and maintain large genomic DNA fragments in both Escherichia coli and Agrobacterium tumefasciens were developed in Arabidopsis (Liu et al. 1999) and wheat (Liu et al. 2000). However, BIBAC and TAC clones that contain fragments of repetitive or single-copy DNA larger than 100 kb were shown to be unstable in Agrobacterium, including recA deficient strains (Song et al. 2003). The same results were obtained for a 83,102 bp Arabidopsis BIBAC clone also shown to be unstable in Agrobacterium (Chang et al. 2011) and for a 92-kb BIBAC clone that was shown to be subject to long stretches of deletions in Agrobacterium (Nakano et al. 2005). In the same publication, these authors analyzed transgenic rice plants produced by Agrobacterium-mediated transformation with a large T-DNA insert containing a 92-kb region of the wheat genome by fluorescence in situ hybridization on extended DNA fibers (fiber FISH). Their results suggest that the large T-DNAs integrated by Agrobacterium-mediated transformation tend to be rearranged in transgenic rice plants (Nakano et al. 2005). On the other hand, Shi et al. (2011) reported that, after being cultured for 96 h in LB medium, maize B73 BIBAC clones were stable in Agrobacterium. Recently, Wang et al. (2015) succeeded in transforming rice with a 164-kbp maize BIBAC clone, but the inheritance and stability of the transgene in the T1, T2 and T3 generations remains unclear. One way to succeed in stable transfer of large fragments into plant genomes without the use of Agrobacterium could be to use biolistic transformation. Ercolano et al. (2004) used this technology to introduce a 106-kb BAC plasmid. The authors used PCR to check that the R1 gene and most of the BAC sequences were present in the resulting transformed plants. However, the plasmid was transferred as a circular molecule, which had to be broken to be inserted, and so there is no evidence—and little likelihood—that the 106-kb fragment was intact. The same conclusions can be drawn for rice, which was transformed with circular BACs containing random genomic DNA (122–157 kbp) or centromere-specific sequences (74–102 kbp) (Phan et al. 2007), and for tobacco, which was transformed with a circular BIBAC containing 83 kb of Arabidopsis genomic DNA (Chang et al. 2011). An alternative method using bioactive beads was applied by Wada et al. (2009) to transfer a ~80-kb circular BAC containing the Hardness (Ha) locus of Aegilops tauschii into the rice genome. Once again, the probability that the large DNA fragment was unbroken is very low because a circular BAC was used for transformation. Large linear DNA fragments of, respectively, 80 and 150 kb or 23 and 50 kb cloned into YAC (yeast artificial chromosome) vectors were successfully used to transform tobacco cells (Mullen et al. 1998), or tobacco plastid DNA (Adachi et al. 2007) using biolistic technology. Despite this success, chimaerism, instability and technical difficulties in the preparation and purification of YAC (Monaco and Larin 1994) have drastically limited the development of this kind of library. So, the transfer of large unbroken DNA fragments into the plant genomes without rearrangements remains a challenge. Recently, we developed a strategy that involves the transfer of dephosphorylated linear fragments (dephosphorylated cassettes) for increased production of transgenic wheat with simple insertions (Tassy et al. 2014). In the present study, we tested the efficiency of the same strategy for the transfer of large DNA fragments into the wheat genome. A 53-kb linear cassette was produced, purified and dephosphorylated to limit concatemerization and to produce more simple integration patterns. This cassette contained a 44-kbp BAC insert originating from A. thaliana to minimize the risk of cross hybridization with wheat genomic DNA, flanked by one selection gene and one reporter gene. Our results showed that transgenic plants harboring the entire linear cassette were generated. The inheritability of the components of the cassette is demonstrated in the T1 and T2 generations.
Materials and methods
Plasmid construction
A vector containing the cassette used for wheat transformation, named pLFg44p, was constructed based on the vector pBelobac11 (NEB-E4154S) in the three following steps:
Construction of the [AscI-gus-NotI-pmi-AscI] cassette
The [AscI-gus-NotI-pmi-AscI] cassette (Supplementary data 1) was constructed using Gateway MultiSite technology (Life technologies). A NotI restriction endonuclease cutting site was inserted between the gus and pmi cassettes to facilitate subsequent cloning of the NotI-NotI Arabidopsis fragment. Construction of the 3′ selection cassette containing the maize ubiquitin promoter, the first intron of the ubiquitin gene, the pmi (manA) coding sequence and the terminator sequence of the nopaline synthase gene of A. tumefasciens is described in Tassy et al. (2014). The central cassette containing the NotI restriction enzyme site was assembled by combining the two following complementary primers 5′ > 3′GGGGACAAGTTTGTACAAAAAAGCAGGCTGCGGCCGCACCCAGCTTTCTTGTACAAAGTGGT and 5′ > 3′ CCCCTGTTCAAACATGTTTTTTCGTCCGACGCCGGCGTGGGTCGAAAGAACATGTTTCACCA. The 5′ cassette containing the gus reporter gene was constructed in the same way as the pmi gene. The Gateway MultiSite (Life Technology) combination of the three cassettes was performed following the manufacturer’s instructions (Supplementary data 1). The resulting [AscI-gus-NotI-pmi-AscI] construction was cloned in the Gateway pDEST R4-R3 vector (Life Technology).
Subcloning of the [AscI-gus-NotI-pmi-AscI] cassette on a modified pBeloBac11 vector
First, the pBeloBac11 vector (NEB-E4154S) was engineered to replace the unique NotI cloning site by an AscI site. The original pBeloBac11 plasmid was linearized using NotI. The double strand DNA resulting from the hybridization of the following synthetic oligonucleotides [5′NotAsc] (5′GGCCATGAGGCGCGCCCT3′) and [3′NotI AscI] (5′GGCCAGGGCGCGCCTCAT3′) was cloned into the NotI site. This fragment included an AscI site (bold nucleotides) and the four protruding bases GGCC, that were cohesive with NotI GCGGCC ends but that did not reconstitute the NotI site. Ligation reaction was performed using T4 DNA ligase supplemented with NotI nuclease to avoid self-ligation of the NotI digested pBeloBac11 plasmid. The resulting vector was named [pBeloBac11_Asc].
The modified plasmid was introduced into E. coli cells (DH10b Electromax Invitrogen), by electroporation, using a BioRad MicroPulser according to the instruction manual. Transformed cells were selected on LB-agar plates containing 12.5 µg/ml of chloramphenicol. Plasmid DNA was extracted from recombinant colonies using the Qiagen Miniprep Kit. AscI and NotI digests allowed us to check for the presence of the AscI site (linearization of the plasmid) and the absence of NotI site (undigested plasmid).
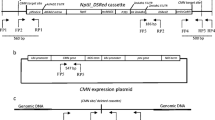
Second, the [AscI-gus-NotI-pmi-AscI] cassette was cloned into the [pBeloBac11_Asc] plasmid. pDEST R4-R3 vector containing the [AscI-gus-NotI-pmi-AscI] cassette was submitted to AscI digest (1 h, one unit of enzyme per µg of plasmid DNA). Restriction fragments of 2.5 kb (vector) and 8.9 kb (cassette) were separated by electrophoresis on 1% agarose gel (50 mA, 5 V/cm). An agarose plug containing the 8.9-kb fragment corresponding to the cassette was cut onto the gel. DNA was recovered using the QIAquick Gel Extraction Kit (Qiagen) according to the instruction manual. [pBeloBac11_Asc] vector was digested by AscI and dephosphorylated as described in Sambrook el al. (1989). Finally, the purified cassette was cloned into the dephosphorylated vector. Ligation reaction was introduced into E. coli DH10b Electomax (Invitrogen). Transformed cells were selected on LB-agar medium containing chloramphenicol 12.5 µg/ml. Plasmid DNA was extracted using BAC or BiBAC miniprep DNA protocol (Sambrook et al. 1989) and screened for the presence of the cassette inserted into the [pBeloBac11_Asc] vector by XhoI digest and fingerprinting. The resulting vector named pLFgp (pLF for plasmid Large Fragment, g for gus and p for pmi) is presented in Fig. 1a.

Construction of the cLFg44p cassette. a pLFgp (plasmid Large Fragment p mi g us) was based on pBeloBac 11_Asc vector. It contained a cassette including a unique NotI site flanked by pmi and gus reporter genes and two AscI sites. The two reporter genes were under the control of maize ubiquitin promoter followed by the first intron of the maize ubiquitin gene. The unique NotI enzymatic site located between the two reporter genes will be used for subsequent subcloning of the Arabidobsis genomic fragment. The two AscI flanking sites will be used for cassette release. This cassette was constructed using the MultiSite Gateway technology. b pLFg44p plasmid. The 44-kbp Arabidopsis BAC insert was released by NotI digest, purified and cloned into the unique NotI site of the pLFgp plasmid. c The final cLFg44p cassette was released from the pLFg44p plasmid by AscI digest. Bold lines under the cassette represent the different probes used for Southern analysis. Gus and pmi probes were obtained by PCR amplification on pLFg44p plasmid using previously described primers. XhoI digest of the pLFg44p plasmid generated a fingerprint containing a unique XhoI-XhoI fragment named 10 kpb Ath probe. This fragment was recovered by gel-elution after agarose gel electrophoresis
Purification and subcloning of the 44-kb Arabidopsis genomic fragment
The BAC DNA (ref. F12K22) was provided by the Arabidopsis Biological Resource Center. Arabidopsis inserts were originally cloned into a pBeloBACKan vector as partial EcoRI fragments. We previously checked that there were no specific genes or sequences in the insert and no NotI enzymatic site. Using the NCBI-BlastN program (Altschul et al. 1990), we also checked that there is no homology between this Arabidopsis sequence and the wheat genome to avoid false positives by cross hybridization. BAC DNA was extracted using the QIAGEN Large-Construct Kit (ref. 12462) following the manufacturer’s instructions. DNA was submitted to NotI enzymatic digestion for 3 h at 37 °C using one unit of enzyme per µg of DNA. BAC vector (pBeloBacKan, 8774 bp) and 44-kb Arabidopsis fragments were separated by pulse field gel electrophoresis on a Bio Rad CHEF Mapper in 0.5X TBE buffer, with the following setting: auto algorithm 5–50 kb—1% agarose gel—14 °C—20 h 30 min. The Arabidopsis DNA fragment was cut into the gel, recovered by electro-elution and dialysis as described in Peterson et al. (2000). The vector pLFgp was digested by NotI in the same conditions as for the BAC DNA and dephosphorylated. The 44-kb fragment was ligated on the pLFgp vector as described in Peterson et al. (2000). The ligation reaction product was cloned on DH10b Electromax E. coli (Invitrogen). The presence of the 44-kb insert was checked by XhoI (fingerprint), NotI and AscI digests. One out of 96 colonies gave the right digest profile for the three endonucleases. The resulting plasmid was named pLFg44p (Fig. 1b).
Production of cLFg44p DNA cassette
The clone containing pLFg44p plasmid was cultured overnight in 500 ml bottles containing 250 ml of LB medium supplemented with 12.5 ug/ml chloramphenicol. Plasmid pLFg44p was recovered and purified using the QIAGEN Large-Construct Kit (ref. 12462), according to the instruction manual. AscI restriction enzyme digest was performed overnight using one unit of enzyme per µg of plasmid DNA. The resulting 53-kb cassette cLFg44p (Fig. 1c) was separated from the 6.9-kb plasmid by electrophoresis on pulse field gel and purified as previously described. Finally, the cassette was dephosphorylated as described in Tassy et al. (2014) and the concentration was adjusted to 30 ng/μl.
Plant material and transformation protocol
Triticum aestivum cv. Bobwhite S26 was used for all experiments. Transformation experiments and plantlets regeneration were performed as described in Tassy et al. (2014), according to Pellegrineschi et al. (2002) and Wright et al. (2001).
Histochemical GUS assay
Expression of the gus gene was checked using the X-Gluc substrate with young leaves of transgenic plant as described in Jefferson (1987). Leaf tissues were cut into small pieces and incubated for 24 h at 37 °C in X-Gluc buffer. Chlorophyll was extracted from the tissues by incubation in 70 °C ethanol at 37 °C to be able to see the blue staining of the transformed tissues.
PCR analysis
Wheat genomic DNA was isolated from young leaves using a modified cetyltrimethylammonium bromide (CTAB) method (Tassy et al. 2006). The gus gene was detected by PCR amplification using 50 ng of total genomic DNA as template and Taq DNA polymerase (Q-BIO gene) following the manufacturer’s instructions, using the primers [GUS A forward 5′ TGC CAG GCA GTT TTA ACG ATC 3′]–[GUS B reverse 5′ AAT CAG GAA CTG TTC GCC CTT 3′], for the GUS assay, and [PMI-1 forward 5′ GCA CAG CCA CTC TCC ATT CA 3′]–[PMI-2 reverse 5′ ATC GGA GTT TGC CAT CAC TTC 3′] for the gus and pmi assay, respectively.
The PCR reaction mixture (50 µl) contained 1X Taq polymerase buffer (Qiagen) containing 1.5 mM MgCl2, 150 µM of each dNTP, 20 pmol of each primer, and one unit of Taq polymerase (Qiagen). PCR was performed using the MJ research PCR thermocycler with one cycle at 94 °C for 3 min, 34 cycles at 94 °C for 45 s, 62 °C for 45 s, 72 °C for 90 s and a final extension step of 72 °C for 5 min for the pmi assay and with one cycle at 94 °C for 3 min, 30 cycles at 94 °C for 30 s, 62 °C for 30 s, 72 °C for 1 min and a final extension step of 72 °C for 5 min for the gus assay. The 543-bp and 710-bp PCR products for pmi and gus genes, respectively, were detected by electrophoresis on a 2% agarose gel and visualized by ethidium bromide staining.
Southern blot analysis
The DNA probes used for Southern blot analysis were produced as follows: gus and pmi probes (Fig. 1c) were generated by PCR amplification using primers GUSA/GUSB; PMI1/PMI2 described above and Gateway pDEST R4-R3 vector containing the [AscI-gus-NotI-pmi-AscI] cassette as DNA template. The PCR products were purified after agarose gel electrophoresis, using the QIAquick Gel Extraction Kit (Qiagen ref. 28704). The Arabidopsis 10-kb probe (Fig. 1c) was produced by XhoI digest of the F12K22 Bac and the 10-kb fragment was recovered after electrophoresis on 1% agarose gel. Purification was performed as described above. The cLFg44p cassette (Fig. 1c) was used as probe for in situ hybridization.
Plant genomic DNA was isolated from young leaves using a modified CTAB protocol (Tassy et al. 2006). Aliquots (15 µg) were digested for 6 h using 10 units per µg of genomic DNA of the appropriate restriction enzyme. Restriction fragments were then separated by electrophoresis in 1% agarose gel in 1X TAE (Tris–Acetate EDTA) buffer, or by pulse field gel electrophoresis as previously described, and blotted by alkaline/saline transfer on a nucleic acid transfer membrane (Amersham hybond N+). Probes were labeled with 32PdCTP, using random hexamer primer method (Megaprime DNA Labeling System GE healthcare ref RPN1606). Southern hybridization was carried out as previously described in Barret et al. (1998).
FISH analysis
Root preparation
Root tips for cytological studies were obtained from T1 plants. T1 seeds were first sterilized in a 5% sodium hypochlorite solution and then germinated on moist filter paper in a Petri dish for 24–48 h at room temperature. The seedlings were then left at 4 °C for 48 h, and at room temperature for 24 h. Young roots were harvested when they were about 2–3 cm long. They were pre-treated in ice water for 24 h to accumulate mitotic cells in metaphase before fixation in 3/1 ethanol/glacial acetic acid solution. The material was fixed at least 48 h before preparation of microscope slides.
Slide preparation for in situ hybridization
The best slides were selected by phase contrast microscopy. They were prepared as described in Mirzaghaderi (2010), Mirzaghaderi et al. (2010).
DNA probes
The pLFg44p cassette labeled with Biotin Nick Translation Kit (Roche 11,745,824,910) according the manufacturer’s instructions was used as FISH probe. To identify A, B or D genomes, two specific probes labeled with fluorescein were added: clone TAS40 (NCBI accession number DX373040) containing a 1-kb fragment from the 3B chromosome from Triticum aestivum cv Courtot and specific to the B genome (Paux et al. 2006), and clone pAs1 probe containing a 1-kb fragment from Aegilops squarrosa (Rayburn and Gill 1986) specific to the D genome.
Fluorescence in situ hybridization
FISH was performed as described in Schwarzacher and Heslop-Harrison (2000).
Results and discussion
Functionality of gus and pmi genes on the [AscI-gus-NotI-pmi-AscI] cassette
Before starting the final construct with the 44-kbp Arabidopsis insert, we checked the functionality of both gus and pmi genes of the [AscI-gus-NotI-pmi-AscI] cassette. To this end, 200 immature wheat embryos were bombarded with the [AscI-gus-NotI-pmi-AscI] cassette and 200 more were bombarded without DNA as control. The embryos were first analyzed for transient gus expression. All the embryos bombarded with the cassette presented GUS phenotype 3 days later, we could thus conclude that the gus reporter gene cassette was functional. Ten plantlets were regenerated by in vitro culture on mannose selective medium, and nothing on control experiment, indicating the functionality of the pmi expression cassette. Among the 10 plantlets, four presented stable GUS expression in young leaves and roots. Recovery of plants expressing both reporter genes indicated that the gus and pmi genes in the [AscI-gus-NotI-pmi-AscI] cassette were functional. The plasmid containing the [AscI-gus-NotI-pmi-AscI] cassette was used to clone the BAC insert of Arabidopsis (NotI-NotI fragment) in the NotI restriction site to obtain the pLFg44p plasmid (Fig. 1).
Cassette cLFg44p production process: quality checking
AscI digest of the pLFg44p plasmid liberated the AscI-AscI cLFg44p cassette (Fig. 1c). The quality of this cassette was checked by agarose gel electrophoresis (Fig. 2). We obtained a single band of the right size (53 kbp), without low-molecular weight degraded DNA, thereby demonstrating that it is possible to obtain large pure intact DNA fragments using PFEG technology combined with electro-elution dialysis.

Gel electrophoresis of the purified 53-kbp cassette cLFg44p. Lambda DNA mono-cut ladder is a high molecular weight ladder (New England Biolabs)
Wheat transformation with cLFg44p cassette and functional characterization of T0 transgenic plants
Wheat transformation experiments were performed with the cLFg44p cassette. The total size of this linear, dephosphorylated cassette was 53kbp. We chose to transfer the linear cassette instead of circular plasmid to avoid random breaking of the insert structure.
We first bombarded 800 immature embryos in two independent transformation experiments. Twenty-four plantlets were regenerated on mannose selective medium. The transformation efficiency (the number of plantlets regenerated to the number of embryos shot) was 3%, which is similar to our usual results. In our hands, 98% of the transgenic wheats obtained after mannose selection were transgenic for the pmi gene (data not shown), so we postulate that all the 24 transgenic events were PMI positive (right border of the cassette). Analysis by GUS assay shown that nine out of the 24 transgenic events (38%) presented GUS expression (left border of the cassette). After the first round of analysis, nine plants were identified that presented evidence for the presence of the two extremities gus and pmi of the transferred cassette, which was a good indicator of the presence of the 44-kbp Arabidopsis genomic fragment. Two of these plants died at the end of in vitro culture process. The seven remaining plants were tagged T0_I to T0_VII and subsequently underwent molecular analysis. A third batch of transformation experiment was performed 6 months later to validate our first transformation results (Supplementary data 2).
Molecular analysis of T0 transgenic events
Genomic DNA of the seven selected T0 plants was extracted from young leaf tissue, digested by three different restriction enzymes and analyzed by Southern blot to characterize the integration profiles of the DNA cassette in the transgenic plants.
Genomic T0 DNA was first digested by XhoI and hybridized with the cLFg44p cassette as probe. Fingerprints obtained for T0 plants were compared to the fingerprint generated by the cLFg44p cassette itself. The theoretical profile of a complete copy insertion was attempted to present the 9 XhoI internal restriction fragments plus two supplementary bands corresponding to the extra-fragments of unknown size hybridized by the borders of the probe (Fig. 3; Table 1). Five out of the seven T0 plants showed a correct fingerprint. In the second step, the number of copies of the transgene was estimated. Genomic DNA from the T0 plants was digested with HindIII and hybridized by gus probe (Fig. 1 for gus probe and HindIII restriction map, and Fig. 3 for Southern analysis). In this configuration, each band resulted from an insertion event of the cassette, depending on the position of the first HindIII site on the flanking genomic DNA. Therefore, the number of bands was directly correlated with the minimum number of copies of the cassette inserted into the wheat genome. T0 plants I, II, VI and VII presented a single band on Southern autoradiography (Fig. 3; Table 1), suggesting that a single copy of the cLFg44p cassette was inserted into the genome of these plants. These results are consistent with the results obtained by Fu et al. (2000), Yao et al. (2006) and Tassy et al. (2014) showing that the use of linear DNA led to a small number of copy insertions. In a second experiment, the same Southern blot was hybridized by the pmi probe (Fig. 1) and only T0 plants VI and VII presented a single band pattern (data not shown). Taken together, these experiments suggest that T0 plants number VI and VII had single-copy insertions of the cLFg44p cassette.

a Southern blot analysis of T0 plants digested by XhoI using the whole cLFg44p cassette as probe. Plant genomic DNA was digested by XhoI and restriction fragments were separated by agarose gel electrophoresis. XhoI digest of the cLFg44p cassette was used as control. Southern experiments were performed using the whole cLFg44p cassette as probe. Size of the XhoI restriction fragments of cLFg44p are indicated on the restriction map. T0 plants with correct fingerprint are tagged with an asterisk. Arrows indicate the two extra-fragments of event VII. b Southern blot analysis of T0 plants for insertion copy number. Genomic DNA from T0 plants was digested by HindIII endonuclease. Restriction fragments were separated by agarose gel electrophoresis, transferred onto a membrane and hybridized by a gus probe. The number of bands is directly correlated with the number of copies of the cassette inserted into the wheat genome
The intactness of the 53-kbp cassettes inserted into T0 plants was assessed by probing genomic DNA digested by AscI with a 10-kb Arabidopsis probe (Fig. 1). As AscI did not cut into the cassette (Fig. 1), we attempted to obtain a fragment larger than the entire cLFg44p DNA. AscI-digested T0 genomic DNAs were separated using pulse field gel electrophoresis to allow efficient separation of large DNA fragments. T0 plants VI and VII have a single band (Fig. 4). These two bands were larger than the cLFg44p control. There was no detectable hybridization signal for T0 plants I–V, which was not consistent with the fingerprints observed for these T0 plants on XhoI Southern blot (Fig. 3). We hypothesize that the Southern PFG electrophoresis was underexposed and that the bands of the T0 events I–V remained under the detection limit, which could be the consequence of the large size of the wheat genome (16 Gbp) that limits the number of copies on the blot. The same results were observed for the third transformation batch (Supplementary data 2). Moreover, PFG electrophoresis was used to separate large fragments that were difficult to handle and to transfer on the blot, which could limit their level of detection. Results obtained using molecular analyses are summarized in Table 1.

Southern analysis of DNA from T0 plants digested by AscI and probed with 10-kb Arabidopsis insert. Large DNA fragments generated by the 8-bp site AscI endonuclease were separated by PFGE
Two out of the seven T0 plants (events VI and VII) gave correct profiles in all the Southern analyses. Two additional fragments (~5 and ~6 kbp) were clearly visible on event VII. The size of these fragments, which were larger than the borders of the cassette, and their specificity to the particular event VII, was consistent with the hypothesis of extra-border fragments. According to these data, we postulated that a single intact copy of the cLFg44p cassette was inserted in these two plants. Subsequent analysis was performed to check the heritability and the stability of the insertion in the T1 generation for events VI and VII.
Stability and segregation of the 53-kb insert
Primary transformed plants (T0) were grown in the glasshouse and self-pollinated to produce T1 seeds for genetic and functional analysis of the transferred cassette. Six T1 seeds of each VI and VII T0 event were sown. Inheritance and structural stability of the transgene on T1 progeny were checked by Southern analysis (Fig. 5). The Southern hybridization profiles of T1 plants were the same as those of the corresponding T0 plants, indicating that the large fragment was stably inherited in the T1 generation. Moreover, the two additional ~5 and ~6-kbp bands identified in plant T0VII that could correspond to extra-border fragments were clearly detected in T1VII progeny (Figs. 3, 5, fragments A and B, and Table 2), showing that the integration site was stable.

Fragment C (Fig. 5; Table 2) was not detected in the T0 VII plant but was clearly detected in plants T1VII-1 and T1VII-4, with no detectable modification of the original T0 pattern. Even if the initial XhoI profile remained intact, we cannot exclude the possibility that fragment C corresponded to duplication or rearrangement of the cassette fragment. Fragments D and E (Fig. 5; Table 2) were faintly detected in T0 VII plants and in the T1 progeny, and could correspond to artefactual, low specific hybridizations, or to additional borders fragments indicating two independent insertion events. For event VI, a faint fragment F was detected on the T0 VI fingerprint, clearly detected on T1 VI-1, VI-3, VI-5 and VI-6 plants, and not detected on plant VI-4 (Fig. 5; Table 2). This fragment appeared to be in segregation, and could correspond to an extra-border fragment. Indeed, the hypothesis of a single-segregating fragment was not compatible with the hypothesis of a single integration site (in which case, the entire fingerprint would have disappeared from the T1 VI-4 plant), indicating that the entire cassette could have been integrated in two or more loci. Functional analysis by GUS assay showed that gus expression was inherited in T1 progeny (data not shown). We checked also that the intact cassette fragment was present in T1 VI-3, -5 and -6 plants (supplementary data 3).
In conclusion, even if some faint additional bands were detected by Southern analysis, the profile generated by XhoI in T0 plants was definitely transmitted to the T1 progeny, confirming the stability of the integrated fragment. In addition, we confirmed these results by gus and pmi PCR analysis, and by histochemical GUS assay on T2 progeny of VI and VII T1 lines (Supplementary data 4).
Cytogenetic structural analysis of T1 progeny by fluorescence in situ hybridization (FISH)
T1 plants of the event VI were analyzed by FISH using the cLFg44p cassette as probe. Two loci were detected by FISH, one on chromosome 4B and one on chromosome 6B. On chromosome 4B insertion occurred on the telomere, even though on chromosome 6B, insertion occurred in the middle of one arm of the chromosome (see Fig. 6 for plant T1 VI-6 and supplementary data 5). FISH performed on individual plants of the T1 VI population showed that the two loci segregated independently (Supplementary data 5 and Table 2). Interestingly, the T1 plant VI-4 was detected as potentially null segregant for the 6B loci because the hybridization signal was only detectable on the two 4B chromosomes on a complete mitotic plate. This plant consequently showed a single insertion of the cassette in the genome, and showed a complete fingerprint of the cassette on XhoI Southern blots (Fig. 5), indicating that that the insertion contained the complete cassette with no major rearrangements. Taken together, these data suggest that the T1 plants VI-4 contained intact insertion of the entire cLFg44p cassette on chromosomes 4B. Fragment F, which segregated in XhoI Southern blot (Fig. 5), was not detected in plant VI-4, suggesting that this fragment could correspond to an extra fragment of the transformation event located on chromosome 6B (Table 2).

FISH analysis of T1 plant VI-6. Red spots represent the cLFg44p biotin labeled probe. Green spots represent probes specific to genome B (TAS43) or D (PAS1), labeled with fluorescein (color figure online)
FISH analysis of the progeny of event VII also showed two insertion loci on chromosomes 2B and 2D (Supplementary data 6). Plant T1 VII-6 was a null segregant for the 2B locus, and consequently presented a single insertion on chromosome 2D. XhoI Southern showed a complete fingerprint of the cassette with no major rearrangements. These data suggest that plant T1 VII-6 contained a complete and intact cassette inserted on chromosome 2D. Fragments D and E were missing on XhoI Southern only in plant T1 VII-6, suggesting that these two fragments were extra-fragments of the insertion event located on chromosome 2B. In the same way, fragments A and B in Fig. 5 could correspond to extra-border fragments of the insertion located on chromosome 2B. The reason for the presence of fragment C in T1 plants VII-1 and VII-4, and its absence in T0 VII plant remain unclear. One possible explanation is that a rearrangement or a partial duplication of the integrated cassette occurred, but the probability that it occurred twice in independent gametes is small. Another possible explanation involves incomplete digest by XhoI of the DNA of the two T1 plants VII-4 and VII-6 that could generate one supplementary fragment. Another technical explanation could be that this fragment was difficult to detect at the heterozygous stage.
FISH analysis of the T1progeny of two other transgenic events I and III detected insertions at one and two loci, respectively (data not shown). These insertions were located on three different chromosomes, indicating that the cassette was randomly inserted in the genome. Concerning the number of insertion loci, FISH analyses detected two independent insertion loci for events III, VI and VII, and one for event I. In a previous work (Tassy et al. 2014) made with gene cassettes of about 3 kbp, we detected only 13% of the transgenic events that segregated in a non-Mendelian fashion, which could indicate multi-locus insertions. Using Agrobacterium, Cheng et al. (1997) found 32% (16/50) of aberrant segregation and Stoger et al. (1998) found 20% (14/70) using bombardment. Even if we checked only four independent events, it was surprising to find 75% of the plants with two independent insertion sites of the cassette. Two hypotheses offer a possible explanation. First, we postulate that FISH is a more powerful method to detect multi-insertions than Southern or genetic analyses on segregating populations. Southern detection by fingerprinting (Figs. 3, 5) of multi-locus insertions is based on the detection of extra-border fragments, two for one locus, four for two loci, but these extra-fragments hybridize with the radioactive probe only on part of their sequence and are consequently very often poorly detected on the autoradiography. It is also very difficult to use Southern analysis hybridized with a cassette-end probe (in our case the gus or pmi probe in Fig. 1) to distinguish between a duplicated insertion at the same locus and at an independent locus, and also to distinguish between one or more fragments (Fig. 3) when only very faint bands are present on the autoradiography. In the case of genetic analysis, the goal is to detect the statistical difference between segregation of one locus (12/16–4/16) and two loci (15/16–1/16), which for a \(\chi^{2}\) test to have 90% power to detect a significant difference (with alpha = 5%), requires analyzing more than 500 plants. Using a large BAC fragment as probe and FISH, we succeeded in identifying plants harboring two insertion sites, each at the homozygous or heterozygous stage. FISH analysis has also been performed on wheat using shorter probes like cDNA probes (Danilova et al. 2014). Under the first hypothesis, we postulate that in many cases the number of independent insertions is underestimated because the technologies used for the analysis of transgenic plants fail to detect simple insertions at different loci. In our work, we detected four independent events using molecular analysis, but after FISH analysis, seven independent transgenic loci were found to be present in T1 progeny. The second hypothesis is that transformation with large DNA fragments improves multi-locus insertions. The size of the DNA molecule that could potentially form a secondary structure may trigger alternative pathways for DNA integration, which, in turn, could integrate single copies of the DNA at different loci instead of one or more copies at a single location. Validation (or invalidation) of this hypothesis requires new experiments.
After segregation in T1, the three heterozygous mono-locus events tested by Southern fingerprinting (T1 plants VI-1, VI-4 and VII-6) had the correct profile, suggesting the cassette was very often integrated at the two loci with no rearrangement. The two T0 events VI and VII that could have been tested by PFG Southern showed no HMW hybridization fragment to suggest a multi-copy integration locus. Taken together, these results show that, at least for the three T1 plants VI-1, VI-4 and VII-6, a single copy of the cassette was integrated at a single locus, with no detectable rearrangement.
Conclusion
We have demonstrated that biolistic technology can be used to transfer large intact DNA fragments into the wheat genome. Three T1 lines showing a single locus/simple copy insertion of a complete cLFg44p cassette were generated in our experiments, demonstrating the advantages of biolistic techniques for the transfer of large undisturbed fragments of DNA into the wheat genome. Moreover, only DNA of interest was transferred, with no other undesirable sequence, and with no detectable DNA rearrangements due to the use of Agrobacterium. The transferred fragment was correctly transmitted to the progeny, and expression of the gus reporter gene was detectable in T1 and T2 plants. We have also shown that it is very difficult to accurately estimate the number and the integrity of copies of a transgene using only molecular biology techniques, especially in plants with a very large genome like wheat. In this case, the use of FISH as a complement to molecular analysis can provide more precise information.
Our results show that biolistics is one of the best technologies for the introduction of undisturbed DNA fragments into the wheat genome, for example wheat fragments to generate cisgenic (Holme et al. 2013) wheats with no foreign DNA.
Our work also provides evidence that using biolistic technology makes it possible to transfer large undisturbed DNA fragments into the nucleus of the wheat cell. This paves the way for interesting CRISPR/Cas9-mediated targeted integration (Wang et al. 2014) of large DNA fragments containing, for example, clusters of disease resistance genes or new metabolic pathways for synthetic biology. In the same way, CRISPR-Cas9-assisted homologous recombination (Li et al. 2013) would require long stretches of DNA as a donor for the replacement of a complete allele, for example a gene and its complete regulations. Moreover, the DNA fragments, coated on gold beads, are probably supplied to the nucleus at much higher concentrations than by Agrobacterium-mediated transformation, thereby providing a large quantity of matrix for further integration or recombination events.
Author contribution statement
PB, CF, MB and AP conceived and designed the research. AP conducted the experiments. GG provided the FISH data. CT developed GATEWAY tools for cassette construction. AP and PB analyzed the data and wrote the manuscript. All the authors read and approved the manuscript.
References
Adachi T, Takase H, Tomizawa K (2007) Introduction of a 50 Kbp fragment into the plastid genome. Biosci Biotechnol Biochem 71(9):2266–2273
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ (1990) Basic local alignment search tool. J Mol Biol 215:403–410
Barret P, Delourme R, Renard M, Domergue F, Lessire R, Delseny M, Roscoe TJ (1998) A rapeseed FAE1 gene is linked to the E1 locus associated with variation in the content of erucic acid. Theor Appl Genet 96:177–186
Chang Y, Chuang H, Meksem K, Wu F, Chang C, Zhang M, Zhang H (2011) Characterization of a plant-transformation-ready large-insert BIBAC library of Arabidopsis and bombardment transformation of a large-insert BIBAC of the library into tobacco. Genome 54:437–447
Cheng M, Fry JE, Pang S, Zhou H, Hironaka CM, Duncan DR, Conner TW, Wan Y (1997) Genetic transformation of wheat mediated by Agrobacterium tumefasciens. Plant Physiol 115:971–980
Danilova TV, Friebe B, Gill BS (2014) Development of a wheat single gene FISH map for analyzing homoeologous relationship and chromosomal rearrangements within the Triticeae. Theor Appl Genet 127:715–730
Ercolano MR, Ballvora A, Paal J, Steinbiss HH, Salamini F, Gebhardt C (2004) Functional complementation analysis in potato via biolistic transformation with BAC large DNA fragments. Mol Breed 13:15–22
Frary A, Hamilton CM (2001) Efficiency and stability of high molecular weight DNA transformation: an analysis in tomato. Transgenic Res 10:121–132
Fu X, Duc LT, Fontana S, Bong BB, Tinjuangjun P, Sudhakar D, Twyman RM, Christou P, Kohli A (2000) Linear transgene constructs lacking vector backbone sequences generate low-copy-number transgenic plants with simple integration patterns. Transgenic Res 9:11–19
Gerland P, Raftery AE, Seveikova H, Li N, Gu D, Spoorenberg T, Alkema L, Fosdick BK, Chunn J, Lalic N, Bay G, Buettner T, Heilig GK, Wilmoth J (2014) World population stabilization unlikely this century. Science 346:234–237
Hamilton CM (1997) A binary-BAC system for plant transformation with high-molecular-weight DNA. Gene 200:107–116
Hamilton CM, Frary A, Lewis C, Tanksley SD (1996) Stable transfer of intact high molecular weight DNA into plant chromosomes. Proc Natl Acad Sci 93:9975–9979
Holme IB, Wendt T, Holm PB (2013) Intragenesis and cisgenesis as alternatives to transgenic crop development. Plant Biotechnol J 11:395–437
Hu Y, Papagerakis P, Ye L, Feng JQ, Simmer JP, Hu JCC (2008) Distal cis-regulatory elements are required for tissue-specific expression of enamelin (Enam). Eur J Oral Sci 116:113–123
Jefferson RA (1987) Assaying chimeric genes in plants: the GUS gene fusion system. Plant Mol Biol Rep 5:387–435
Li J-F, Norville JE, Aach J, McCormack M, Zhang D, Bush J, Church GM, Sheen J (2013) Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nature 31:688–691
Liu YG, Shirano Y, Fukaki H, Yanai Y, Tasaka M, Tabata S, Shibata D (1999) Complementation of plant mutants with large genomic DNA fragments by a transformation-competent artificial chromosome vector accelerates positional cloning. Proc Natl Acad Sci 96:6535–6543
Liu Y, Nagaki K, Fujita M, Kawaura K, Uozumi M, Ogihara Y (2000) Development of an efficient maintenance and screening system for large-insert genomic DNA libraries of hexaploid wheat in a transformation-competent artificial chromosome (TAC) vector. Plant J 23(5):687–695
Mirzaghaderi G (2010) Simple metaphase chromosome preparation from meristem root tip cells of wheat for karyotyping or in situ hybridization. Afr J Biotechnol 9(3):314–318
Mirzaghaderi G, Karimzadeh HS, Hassani M, Jalali-Javaran M, Baghizadeh A (2010) Cytogenetic analysis of hybrids derived from wheat and Tritipyrum using conventional staining and genomic in situ hybridization. Biol Plant 54(2):252–258
Monaco AP, Larin Z (1994) YACs, BACs, PACs and MACs: artificial chromosomes as research tools. TIBTECH 12:280–286
Mullen J, Adam G, Blowers A, Earle E (1998) Biolistic transfer of large DNA fragments to tobacco cells using YACs retrofitted for plant transformation. Mol Breed 4:449–457
Nakano A, Suzuki G, Yamamoto M, Turnbull K, Rahman S, Mukai Y (2005) Rearrangements of large-insert T-DNAs in transgenic rice. Mol Gen Genom 273:123–129
Naqvi S, Farré G, Sanahuja G, Capell T, Zhu C, Christou P (2010) When more is better: multigene engineering in plants. Trends Plant Sci 15:1360–1385
Paux E, Roger D, Badaeva E, Gay G, Bernard M, Sourdille P, Feuillet C (2006) Characterizing the composition and evolution of homoeologous genomes in hexaploid wheat through BAC-end sequencing on chromosome 3B. Plant J 48(3):463–474
Pellegrineschi A, Noguera LM, Skovmand B, Brito RM, Velazquez L, Salgado MM, Hernandez R, Warburton M, Hoisington D (2002) Identification of highly transformable wheat genotypes for mass production of fertile transgenic plants. Genome 45(2):421–430
Peterson DG, Tomkins JP, Frisch DA, Wing RA, Paterson AH (2000) Construction of plant bacterial artificial chromosome (BAC) libraries: An illustrated guide. J Agric Genom, p 5. www.ncgr.org/research/jag. Accessed 29 June 2017
Phan BH, Jin W, Topp CN, Zhong CX, Jiang J, Dawe RK, Parott WA (2007) Transformation of rice with long DNA-segments consisting of random genomic DNA or centromere-specific DNA. Transgenic Res 16(3):341–351
Rayburn AL, Gill BS (1986) Molecular identification of the D-genome chromosomes of wheat. J Hered 77:253–255
Saidi S, Rival-Gervier S, Daniel-Carlier N, Thépot D, Morgenthaler C, Viglietta C, Prince S, Passet B, Houdebine LM, Jolivet G (2007) Distal control of the pig whey acidic protein (WAP) locus in transgenic mice. Gene 431:97–107
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory. Cold Spring Harbor, New York
Schwarzacher T, Heslop-Harrison P (eds) (2000) Practical in situ hybridization. BIOS Scientific Publishers Ltd, Oxford, UK
Shi X, Zeng H, Xue Y, Luo M (2011) A pair of new BAC and BIBAC vectors that facilitate BAC/BIBAC library construction and intact large genomic DNA insert exchange. Plant Methods 7:33
Song J, Bradeen JM, Naess SK, Helgeson JP, Jiang J (2003) BIBAC and TAC clones containing potato genomic DNA fragments larger than 100 Kb are not stable in Agrobacterium. Theor Appl Genet 107:958–964
Sparks CA, Jones HD (2014) Genetic transformation of wheat via particle bombardment. In: Henry RJ, Furtado A (eds) Cereal genomics: methods and protoccols, methods in molecular biology, vol 1099. Humana Press, New York
Sparks CA, Doherty A, Jones HD (2014) Genetic transformation of wheat via Agrobacterium-mediated DNA delivery. In: Henry RJ, Furtado A (eds) Cereal genomics: methods and protoccols, methods in molecular biology, vol 1099. Humana Press, New York
Stoger E, Williams S, Keen D, Christou P (1998) Molecular characteristics of transgenic wheat and the effect on transgene expression. Transgenic Res 7:463–471
Tassy C, Feuillet C, Barret P (2006) A method suitable for the conservation of wheat tissue samples at room temperature allowing successive cycles of DNA extraction on the same sample. Plant Mol Biol Rep 24:247a–247f
Tassy C, Partier A, Beckert M, Feuillet C, Barret P (2014) Biolistic Transformation of wheat: increased production of plants with simple insertions and heritable transgene expression. PCTOC 119:171–181
Vasil V, Castillo AM, Fromm ME, Vasil IK (1992) Herbicide resistant fertile transgenic wheat plants obtained by microprojectile bombardment of regenerable embryonic callus. Nat Biotechnol 10:667–674
Wada N, Kajiyama S, Akiyama Y, Kawakami S, No D, Uchiyama S, Otani M, Shimada T, Nose N, Suzuki G, Mukai Y, Fukui K (2009) Bioactive beads-mediated transformation of rice with large DNA fragments containing Aegilops tauschii genes. Plant Cell Rep 28:759–768
Wang Y, Cheng X, Shan Q, Zhang Y, Liu J, Gao C, Qui J-L (2014) Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat Biotechnol 32:947–951
Wang Y, Zeng H, Zhou X, Huang F, Peng W, Liu L, Xiong W, Shi X, Luo M (2015) Transformation of rice with large maize genomic DNA fragments containing high content repetitive sequences. Plant Cell Rep 34:1049–1061
Wright M, Dawson J, Dunder E, Suttie J, Reed J, Kramer C, Chang Y, Novitzky R, Wang H, Artim-Moore L (2001) Efficient biolistic transformation of maize (Zea mays L.) and wheat (Triticum aestivum L.) using the phosphomannose isomerase gene, pmi, as the selectable marker. Plant Cell Rep 20:429–436
Yao Q, Cong L, Chang JL, Li KX, Xiao G, He GY (2006) Low copy number gene transfer and stable expression in a commercial wheat cultivar via particle bombardment. J Exp Bot 57(14):3737–3746
Acknowledgements
This work was supported by INRA grants and the publication was supported by the program Investments for the Future (grant ANR-11-BTBR-0006-GENIUS) managed by the French National Research Agency. We are grateful to Arabidopsis Biological Resource Center for providing the BAC clones. We thank Ludovic Georges for technical help with FISH analyses. Special thanks to the greenhouse team, especially Richard Blanc, for taking care of the plants.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Communicated by Pedro Puigdomenech.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Partier, A., Gay, G., Tassy, C. et al. Molecular and FISH analyses of a 53-kbp intact DNA fragment inserted by biolistics in wheat (Triticum aestivum L.) genome. Plant Cell Rep 36, 1547–1559 (2017). https://doi.org/10.1007/s00299-017-2173-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-017-2173-5