
Abstract
Three long-term nodal tissued cultured Russet Burbank potato clones and nine thaxtomin A-treated regenerant lines, derived from the nodal lines, were assessed for genetic and epigenetic (in the form of DNA methylation) differences by AFLP and MSAP. The treated regenerant lines were originally selected for superior resistance to common scab disease and acceptable tuber yield in pot and field trials. The long-term, tissue culture clone lines exhibited genetic (8.75–15.63% polymorphisms) and epigenetic (12.56–26.13% polymorphisms) differences between them and may represent a stress response induced by normal plant growth disruption. The thaxtomin A-treated regenerant lines exhibited much higher significant (p < 0.05) genetic (2–29.38%) and epigenetic (45.22–51.76%) polymorphisms than the nodal cultured parent clones. Methylation-sensitive mutations accumulated within the regenerant lines are significantly correlated (p < 0.05) to disease resistance. However, linking phenotypic differences that could be of benefit to potato growers, to single gene sequence polymorphisms in a tetraploid plant such as the potato would be extremely difficult since it is assumed many desirable traits are under polygenic control.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Plant tissue culture technologies have proven very valuable in horticulture for maintenance of healthy nuclear stocks and rapid propagation of planting material. Most potato seed propagation systems around the world maintain elite clones in tissue culture. These are used to produce micro or mini tubers in glasshouse culture before seed tuber propagation in the field. Whilst clonal propagation through tissue culture should generate individuals identical to the plant from which they were sub-cultured, studies over the past two decades have shown genetic and epigenetic changes may accumulate over time. These may lead to phenotypic changes in the propagated clones and alterations in agronomic performance (Vazquez 2001; Vazquez and Linacero 2010).
Tissue culture practices involving induction of callus cultures and subsequent regeneration of whole plants have been shown to produce a greater frequency of genetic and epigenetic change than propagation of nodal cuttings (Bordallo et al. 2004; Pacheco et al. 2008; Sharma et al. 2007). This induction of variation has been occasionally used to select regenerant variants of target crop plants with superior cultivar traits. Variable proportions of tissue-cultured plants showing polymorphisms have been described in a number of commercial plant species for example, rye (de la Puente et al. 2008), potato (Bordallo et al. 2004; Joyce and Cassells 2002; Nielsen et al. 2007), banana (Lakshmanan et al. 2007), pea (Kuznetsova et al. 2006) and cocoa plants (Rodríguez-López et al. 2010).
The molecular basis for epigenetic and genetic variability, and their interactions, are now becoming apparent and involves profound effects on transcription, transposable nucleic elements and chromosomal changes (Alvarez et al. 2010; Zilberman and Henikoff 2007). Epigenetic changes occur on several interdependent levels including reversible DNA methylation, histone modifications and chromatin remodelling (Grant-Downton and Dickinson 2005) which can be mutually antagonistic (Zilberman et al. 2008). Changes in DNA methylation are quite stable and are frequently transmitted during meiosis, gametogenesis and mitosis (Saze 2008). Hence, advantageous phenotypic and morphological traits induced by genetic and epigenetic changes in commercially important crop plants have become of great importance as these changes have been linked to disease resistance (Abou-Taleb et al. 2010; Alvarez et al. 2010; Sha et al. 2005). However, it can also become an undesirable phenomenon when the main objective is continual propagation of genetically identical plants for cropping.
Methods to detect genetic and epigenetic changes have become more streamlined and exhaustive with next-generation sequencing; however, amplified fragment length polymorphisms (AFLP) and methylation-sensitive amplified polymorphisms (MSAP) are still a reliable and relatively cheap alternative (Aversano et al. 2007; Bayazit et al. 2007; Bonin et al. 2005; Guthridge et al. 2001; Laurentin and Karlovsky 2007; Peredo et al. 2006) and are well known for producing reproducible genetic markers compared with other methods such as random amplification of polymorphic DNA (RAPD) and simple sequence repeats (SSR) in potato (McGregor et al. 2000).
The MSAP method, a modification of the AFLP technique, can be differentiated by the methylation state indicated by digestion with the isoschizomers HpaII and MspI. These isoschizomers recognise the same tetranucleotide sequence 5′-CCGG but have different sensitivities to the methylation of the cytosines: HpaII will not cut if either of the cytosines are fully methylated, whereas MspI will not cut if the external cytosine is fully- or hemi-methylated; they cannot distinguish between unmethylated and fully methylated cytosines or hemi-methylated internal cytosines (McClelland et al. 1994). Thus, for any given DNA sample two major methylation states at 5′-CCGG sites can be distinguished, methylation sensitive and methylation insensitive. Hence, the methylation percentages obtained by this method will be lower than the total absolute values. In spite of this limitation the MSAP method has been successfully applied in a wide range of studies where alterations in cytosine methylation were detected in regenerants of various plant species (Baránek et al. 2010; Hao et al. 2004; Joyce and Cassells 2002; Matthes et al. 2001).
Russet Burbank originally selected as a mutant of cv. Burbank in 1873 (Stansfield 2006) is one of the most commonly cropped potato cultivars in the world whose tuber traits are ideal for French fry processing. Several agronomically distinct clonal lines of Russet Burbank have been identified since its original selection that suit different cropping regions. In Tasmania the Vancouver clone is the main cropping potato. The original Vancouver clone was divided into five separate lines (RBK1–RBK5) (Fig. 1) which were propagated independently in tissue culture to ensure identification of any aberrant phenotypic changes.

Schematic diagram of the origin of the tissue-cultured Russet Burbank potato parental clone lines and thaxtomin A treated regenerant lines and the experimental outline
Common scab is one of the most economically important diseases affecting potato (Loria et al. 1997). Whilst Russet Burbank is regarded as possessing moderate resistance to this disease (Goth et al. 1985), common scab epidemics frequently occur where disease pressure is high (Wilson 2004; Wilson et al. 1999). In order to improve common scab resistance, clones of Russet Burbank were subjected to a somatic cell selection program to obtain regenerant variants with enhanced disease resistance. During selection, cell cultures were exposed to thaxtomin A, the primary phytotoxin produced by pathogenic Streptomyces spp., known to have an implicit role in infection and common scab disease development (Fry and Loria 2002). Regenerated cell lines were screened for enhanced disease resistance, tuber yield and processing qualities (Wilson et al. 2010).
The aim of this study was to determine the level of genetic and epigenetic changes that have occurred within the parent clones after long-term tissue culture, to compare this to changes resulting from toxin-exposed cell selection.
Materials and methods
Plant DNA extraction
Plant DNA was extracted from 100 mg leaves collected from duplicate sub-cultures of six Russet Burbank toxin-exposed regenerant lines (A168a, A380, TC9M4, TC9M4-7, TC-RB8 and TC10C1) and three parent clonal lines (RBK1, RBK3 and RBK5). All regenerant lines were derived from parent clone RBK5, except for TC10C1 and TC-RB8 (from RBK3) (Fig. 1). These lines had been maintained on MS media and incubated under controlled environmental conditions of 22°C, 16/8 h light/dark cycles using fluorescent light (65 μmol m−2 s−1). Total DNA was extracted using the FastDNA®SPIN kit (MP Bio, Solon, USA). First the tissue was homogenised in the FastPrep FP120 instrument for 40 s at maximum speed with lysis matrix D and purification following the manufacturer’s protocol. DNA concentration was estimated using a Picofluor fluorometer (Promega, Sunnyvale, USA) with a Quant-it™ PicoGreen DNA assay kit (Invitrogen, Carlsbad, USA) and DNA was stored at −20°C.
AFLP and MSAP assays
Amplified fragment length polymorphism reactions were carried out according to the protocol described in (Vos et al. 1995) for the enzyme combination EcoRI/MseI and MSAP reactions were carried out according to the protocol described in (Xu et al. 2000) using the enzyme combinations EcoRI/MspI and EcoRI/HpaII with the exception that 10 pmol of HpaII enzyme was used as it has reduced activity in NEB#4 buffer. Adapters and primers are summarised in Table 1. Genomic DNA (500 ng) was incubated for 4 h at 37°C then 16°C overnight with 5 U of each enzyme (New England Biolabs, Ipswitch, USA) for AFLP (EcoRI/MseI) and MSAP (EcoRI/MspI and EcoRI/HpaII) in a solution containing ×1 NEB #4 buffer (New England Biolabs), 50 ng BSA, 2 U T4 ligase, ×1 T4 ligase buffer containing 1 mM ATP, 1 pmol EcoR-adpaters, 5 pmol Mse-adapters (AFLP) or 5 pmol HM-adapters (MSAP) and made up to 50 μL with sterile distilled water. Reactions were diluted to 250 μL with sterile distilled water and stored at −20°C. Adapter mixes were prepared by adding equimolar amounts of both adapters and heating to 95°C for 5 min and slowly cooled to room temperature.
Pre-selective PCRs were prepared using ×1 Reaction Buffer (Bioline, Tauton, USA) 2 mM MgCl2 1 1 mM dNTPs, 0.3 pmol EcoR-T primer, 0.3 pmol Mse-C primer (AFLP) or 0.3 pmol EcoR-T, 0.3 pmol HM-0 primer (MSAP), 1 U Biotaq polymerase (Bioline) and made up to 25 μL with sterile distilled water. The reaction conditions were 1 × 72°C/3 min, 35 × (94°C/30 s, 56°C/30 s, 72°C/2 min) and 1 × 72°C/10 min. All PCRs were visualised in 1.5% agarose gel electrophoresis with SYBR Safe™ DNA Gel Stain (Invitrogen) in 1× lithium borate buffer. The reaction mixture was diluted 1:10,000 with sterile distilled water and stored at −20°C.
Selective PCRs were prepared using ×1 HotStarTaq Master Mix (Qiagen), 1 pmol of each primer in a combination (Table 2), and 1 μL of the diluted template from the pre-selective PCR and made up to 25 μL with sterile distilled water. All PCRs were again visualised by agarose gel electrophoresis. Reactions were stored at −20°C wrapped in aluminium foil. Ten primer combinations were used for AFLP analyses and nine for MSAP analyses.
PCR products (1 μL) were mixed with 30 μL Sample Loading Solution (Beckman Coulter) and 0.3 μL of 600 base pair (bp) DNA size standard (Beckman Coulter) and analysed using a Beckman Coulter CEQ8000 Genetic Analysis System using the Frag-4 method (injection 2.0 kV 30 s−1, run at capillary temperature 50°C at 4.8 kV for 1 h). Results were analysed using the Beckman Coulter CEQ8000 Fragment Analysis System software. The samples were normalised by using a lower threshold peak height of 5% and results were filtered for peak height <5,000 dye fluorescent units (dfu). Peak data were binned as presence/absence (1/0) with a 1 bp binning threshold.
Data analysis
The results from the fragment analysis for all AFLP selective primer combinations were pooled for all samples into one binary matrix and checked for redundant markers using AFLPOP (Duchesne and Bernatchez 2002). The results from the MSAP fragment analysis for all selective primer combinations of the separate digests (EcoRI/MspI and EcoRI/HpaII) were combined into a binary matrix (1 = fragment presence in EcoRI/MspI digest, 0 = fragment presence in EcoRI/HpaII digest), i.e. the different methylation-sensitive states, full (both strands) methylation of cytosines (1) or the external cytosine is fully- or hemi-(single strand) methylated (0). The methylation-insensitive fragments were not included, i.e. the same fragment in both digests (no methylation) or no fragment in either digest (full methylation of cytosines on both strands or hemi-methylated on the internal cytosine). In both AFLP and MSAP analyses polymorphic fragments for each line were counted as a gain or loss of a unique fragment compared with the parent line it was regenerated from and calculated as a percentage of the total number of fragments overall; for example, clone line A168a had 39 gains and losses of unique fragments compared with the parent (RBK5) in a total of 160 fragments (Table 3). Parent clone lines were compared with one another.
Variation was visualised among the regenerant lines based on genetic and epigenetic polymorphism similarities by principal coordinate analysis (PCoA) and dendogram construction by UPGMA (Unweighted pair-group method with arithmetic averages) and the ANOVA was calculated for each variable between the parental (untreated) and regenerant lines (treated). The similarity matrices were generated using Jaccard’s similarity coefficient. All analyses were performed with XLSTAT v 2010.4.01 (Addinsoft New York, USA) in Microsoft Excel®. Disease severity and yield were recorded from seven field and pot trials conducted in between 2004 and 2008 (Wilson et al. 2010). To test for possible relationships between genetic and epigenetic differences and disease and yield differences, Pearson correlation coefficients were generated between all the similarity matrices and tested for statistical significance against 10,000 random permutations by the Mantel test (Mantel 1967). Multiple linear regression analysis was used to identify any correlation between regenerant lines with respect to disease severity and mean total yield traits as the dependent variables and genetic polymorphisms, methylation-sensitive polymorphisms, ratio of gains/losses of genetic polymorphic loci and ratio of gains/losses of methylation-sensitive polymorphic loci (i.e. if the ratio is >1 there is a net gain in polymorphic sites and <1 is a net loss of polymorphic sites) (Table 3) as the explanatory variables.
Results
AFLP analysis
The ten primer sets used for AFLP analysis generated 185 duplicate fragments from all clone lines in this study. This was reduced to 160 by the removal of redundant loci by AFLPOP. Parent clone lines had much less polymorphisms between them representing genetic changes from long-term nodal culture. Within the three parent clones, RBK1 and RBK3 had a relative net gain of polymorphisms (15.63 and 13.13% respectively), with RBK5 showing net loss of common polymorphic sites. The regenerant lines TC9M4, TC9M4-7 and TC10C1 had the highest number of polymorphisms (28–29%) compared with their respective parental lines. Regenerant lines A168a and A380 had the same amount of polymorphism (324.38%) and similar gains and losses of unique polymorphisms to their parent clone RBK5. Regenerant line TC-RB8 was the only clone with yields greater than its parent (RBK5). This clone has the lowest genetic polymorphisms (20%) with a net loss of polymorphic sites (Table 3). Regenerant line A380 had the phenotype combination of greatest disease resistance of the cell lines studied and reasonable tuber yield and presented a high net gain of polymorphic sites (Table 3) similarly with regenerant A168a, indicating increased genetic polymorphism may have conferred some desirable phenotypic traits.
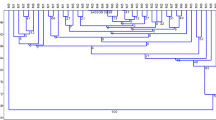
Analysis of variation with AFLP data for all clonal lines illustrated the overall similarities between the parent and clone lines (Fig. 2). The treated regenerant lines clustered with their respect parental lines but there was low overall similarity between the groupings (0.489) and ANOVA indicated the treated lines were significantly different in genetic polymorphisms to the untreated parent lines (p < 0.01). Principal coordinate analyses (PCoA) (Fig. 3) confirmed that the thaxtomin A-treated regenerant lines were genetically similar to their respective parent lines. RBK1 was the least similar to all the other lines and had the highest net % polymorphisms within the parent lines (15.63%) (Table 3). The Mantel test indicated there was no significant correlation (p > 0.05) between the similarity matrices for genetic polymorphisms with disease severity, yield or methylation-sensitive polymorphisms matrices.

UPGMA cluster analysis of AFLP data generated by ten primer combinations for Russet Burbank untreated parent clonal and thaxtomin A treated regenerant lines based on a Jaccard’s similarity matrix

Relationships between Russet Burbank potato untreated parent clonal and thaxtomin A-treated regenerant lines illustrated by Principal coordinate analysis (PCoA) based on a Jaccard’s similarity matrix of AFLP data. Two axes explain 44.23% of the variability
Methylation-sensitive AFLP analysis
The nine primer sets produced 386 reproducible fragments from all the clone lines; this was reduced to 199 by the removal of redundant loci by AFLPOP. The methylation-sensitive polymorphisms (Table 3) were relatively low in the parent lines RBK1 = 12.56%, RBK3 = 26.13% and RBK5 = 23.62%) and represent long-term tissue-culture only effects. The thaxtomin A-treated regenerants had higher methylation polymorphisms compared with their parent lines as expected (45.22–51.76%).
A dendogram for all the clone lines MSAP data (Fig. 4) showed low similarity overall (average 0.510). The PCoA graph showed the overall groupings (Fig. 5). Regenerant line A380 was the most dissimilar to the other lines and A168a grouped closely to the parent lines RBK1 and RBK5, all the other lines grouped together. This illustrates the regenerant lines that were genetically similar were not necessarily epigenetically similar. ANOVA indicated the treated regenerant lines were significantly different to the parent lines with respect to methylation-sensitive polymorphisms (p < 0.05). The Mantel test indicated there was significant correlation (p < 0.05) between similarity matrices for methylation-sensitive polymorphisms and mean disease severity but no significant correlation with the mean total yield similarity matrix.

UPGMA cluster analysis of MSAP data generated by nine primer combinations for Russet Burbank potato untreated parent clonal and thaxtomin A-treated regenerant lines based on a Jaccard’s similarity matrix

Relationships between potato Russet Burbank untreated parent clonal and thaxtomin A-treated regenerant lines illustrated by Principal Coordinate Analysis (PCoA) based on a Jaccard’s similarity matrix of MSAP fragment analysis. Two axes explain 32.94% of the variability
Data analysis
ANOVA of each variable (% genetic polymorphisms, genetic ration, % methylation-sensitive polymorphisms and methylation ratio) indicated % genetic (p < 0.01) and % methylation-sensitive polymorphisms (p < 0.0001) and mean disease severity (p < 0.01) significantly explained the variation between untreated parent line and the treated regenerant lines (Table 3). To test for possible relationships multiple linear regression modelling was performed with all the potato clone lines. Regression indicated disease severity was significantly correlated to all polymorphisms (p < 0.05, R 2 = 0.929), specifically the genetic ratio and methylation-sensitive ratio contributing the highest correlation (R 2 = 0.906). However, there was significant but low correlation with respect to yield (p < 0.05, R 2 = 0.576) with % genetic polymorphism having the highest contribution (R 2 = 0.542). This result was possibly skewed by the regenerant line RC-RB8 as this line had a high yield (\( \overline{x} = 2.45 \)) but low genetic polymorphisms (20%).
Discussion
AFLP and MSAP methods were applied to determine genetic and epigenetic tissue culture-induced variation in long-term, nodal tissue-cultured Russet Burbank potato parent clone lines and recently produced regenerant variants from callus culture that have been selected for superior disease resistance traits. The parent clone lines had been independently sub cultured by nodal cuttings in tissue culture for approximately 40 years. AFLP and MSAP analyses indicated some genetic variation and comparatively low methylation-sensitive polymorphisms have been acquired by separate clonal culture during this time (Table 3). This indicated long-term nodal tissue culture may induce some plant stress responses as normal cellular processes are disrupted, which results in a cascade of genetic and epigenetic instabilities but not enough to produce phenotypic differences in this case, although it has been found in other instances to be quite substantial, even autoploidy (Phillips et al. 1994). Comparatively the thaxtomin A-treated regenerants, selected from RBK5 to RBK3 parent lines 9 years ago, have significant increases in both genetic and methylation-sensitive polymorphisms evident across all the regenerant lines (Table 3). However, the number and net gain and loss of polymorphic sites are variable. Hence, the DNA methylation state is also prone to alterations in response to various stresses and new epialleles can be formed which may allow an adaptive response with the particular stress (Richards 2006). DNA methylation appears to have been the main contributor to acquired enhanced disease resistance in the thaxtomin A-treated potato clone lines and the amount of polymorphic sites gained and lost also appears important. There was no correlation between the similarities found in AFLP analysis and MSAP analysis unlike Guo et al. (2007), Li et al. (2007) who found methylation was significant in regenerants of Condonopsis lanceolata but was correlated to RAPD and ISSR genetic analyses and was not different to that of the donor plant and hence, tissue-culture alone has not contributed to the over-all differences between the clone lines. Li et al. (2007), however, found correlation only between MSAP and S-SAP (Sequence-specific amplification polymorphism) and not between MSAP and AFLP results, as in this study, indicating that intrinsic interrelatedness of genetic and epigenetic stability may exist only in certain genomic regions such as regions flanking retrotransposons. Understanding the causal relationships between DNA methylation and genetic changes and their collected contribution to novel phenotypes is of great importance (Li et al. 2007).
The regenerant line with the greatest enhanced disease resistance (A380; 78% reduction in disease severity compared with parent) and reasonable yield was the only line with an overall reduction in methylation sites compared with its parent line RBK5. Sha et al. (2005) found differential cytosine methylation in adult rice plants compared with seedlings led to differential expression of certain genes which may be associated with adult plant resistance to bacterial blight. Recent research has also shown pathogen-induced DNA methylation changes can occur in Arabidopsis (Pavet et al. 2006) and tobacco (Wada et al. 2004) but the effect on disease resistance is unknown. The treatment with thaxtomin A during cell selection is analogous to exposure to the common scab pathogen S. scabiei as this toxin is the principal virulence factor and may have been responsible for some of the genetic rearrangements and cytosine methylation changes.
Complicating epigenetic variation is genetic variation. The regenerant line A380 was least like the other regenerant lines (Fig. 2) and had the highest gain to loss ratio of genetic polymorphic sites but one of the lowest in methylation-sensitive polymorphism ratio (Table 3). Similarly, the regenerant line A168a also had a high gains to loss ratio of polymorphic sites and had low disease severity but lower yield. The line TC-RB8 had a net loss of genetic polymorphic sites compared with the parent line RBK5 (Table 3). This regenerant has the highest mean disease severity (but still a 43% reduction in disease severity compared to its parent) but showed significant increased yield (137% increase to parent). Pathogen-induced systemic plant signalling triggering DNA rearrangements have previously been found in tobacco (Boyko et al. 2007). Furthermore, Boyko et al. (2007) found increases of global genome methylation a general mechanism against stress in response to pathogen infection but also found locus specific de-methylation was associated with a higher frequency of rearrangements. This was indicated in the thaxtomin A-treated potato clone line A380 which had high genetic polymorphism but a net loss of methylation-sensitive polymorphisms. The same effect was observed when plant pathogen-defence mechanisms were activated by the chemicals 2,6-dichloroisonicotinic acid (INA) or benzothiadiazole (BTH) (Lucht et al. 2002). These findings suggest that increased somatic recombination is also a general stress response in plants. This increased genetic and epigenetic flexibility might facilitate adaptation to stressful environments or events, such as infection, even in long-term, tissue-cultured lines that have limited natural variability, for improved cultivars (Schellenbaum et al. 2008). Induced mutation by exposure to phytotoxins has long been a method for crop improvement (Cassells 1998), but improved genetic methodologies has allowed greater insight into the underlying genetic and epigenetic changes occurring.
Most of the Russet Burbank tissue culture lines had at least one unique genetic marker. More primer combinations may have revealed more unique polymorphisms. However, linking phenotypic differences that could be of benefit to potato growers, to single gene sequence polymorphisms in a tetraploid plant such as the potato would be extremely difficult since it is assumed many desirable traits are under polygenic control (Visser et al. 2009) and differential gene expression (Hegarty and Hiscock 2008). Adding to this complexity is epigenetic effects on those genes and their expression. Despite the importance of the potato as a global food crop and long history of farming many qualitative and quantitative agronomic traits are poorly understood. Extensive sequencing of the AFLP and MSAP fragments and global gene expression analyses may be a valuable strategy to examine genomic regions most affected by genetic and epigenetic changes, progressing towards identification of gene sequences associated with observed phenotypic changes.
References
Abou-Taleb EM, Aboshosha SM, El-Sherif EM, El-Komy MH (2010) Genetic diversity among late blight resistant and susceptible potato genotypes. Saudi J Biol Sci 17:133–138
Alvarez ME, Nota F, Cambiagno DA (2010) Review: epigenetic control of plant immunity. Mol Plant Pathol 11:563–576
Aversano R, Ercolano MR, Frusciante L, Monti L, Bradeen JM, Cristinzio G, Zoina A, Greco N, Vitale S, Carputo D (2007) Resistance traits and AFLP characterization of diploid primitive tuber-bearing potatoes. Genet Resour Crop Evol 54:1797–1806
Baránek M, Křižan B, Ondrušíková E, Pidra M (2010) DNA-methylation changes in grapevine somaclones following in vitro culture and thermotherapy. Plant Cell Tiss Organ Cult 101:11–22
Bayazit S, Kazan K, Gülbitti S, Çevik V, Ayanoğlu H, Ergül A (2007) AFLP analysis of genetic diversity in low chill requiring walnut (Juglans regia L.) genotypes from Hatay, Turkey. Sci Hortic 111:394–398
Bonin A, Pompanon F, Taberlet P (2005) Use of amplified fragment length polymorphism (AFLP) markers in surveys of vertebrate diversity. Methods Enzymol 395:145–161
Bordallo PN, Silva DH, Maria J, Cruz CD, Fontes EP (2004) Somaclonal variation on in vitro callus culture potato cultivars. Hortic Bras 22:1–6
Boyko A, Kathiria P, Zemp FJ, Yao Y, Pogribny I, Kovalchuk I (2007) Transgenerational changes in the genome stability and methylation in pathogen-infected plants: (Virus-induced plant genome instability). Nucleic Acids Res 35:1714–1725
Cassells AC (1998) In vitro-induced mutations for disease resistance. In: Mohan Jain S, Brar DS, Ahloowalia BS (eds) Somaclonal variation and induced mutations in crop improvement. Kluwer Academic Publishers, Dordrecht, The Netherlands, pp 367–379
de la Puente R, González AI, Ruiz ML, Polanco C (2008) Somaclonal variation in rye (Secale cereale L.) analysed using polymorphic and sequenced AFLP markers. In Vitro Cell Develop Biol Plant 44:419–426
Duchesne P, Bernatchez L (2002) AFLPOP: a computer program for simulated and real population allocation based on AFLP data. Mol Ecol Notes 3:380–383
Fry BA, Loria R (2002) Thaxtomin A: evidence for a plant cell wall target. Physiol Mol Plant Pathol 60:1–8
Goth RW, Haynes KG, Young RJ, Wilson DR, Lauer FI (1985) Relative resistance of the potato cultivar Krantz to common scab caused by Streptomyces scabies as determined by cluster analysis. Am J Potato Res 72:505–511
Grant-Downton RT, Dickinson HG (2005) Epigenetics and its implications for plant biology. 1. The epigenetic network in plants. Ann Bot 96:1143–1164
Guo WL, Wu R, Zhang YF, Liu XM, Wang HY, Gong L, Zhang ZH, Liu B (2007) Tissue culture-induced locus-specific alteration in DNA methylation and its correlation with genetic variation in Codonopsis lanceolata Benth et Hook. f. Plant Cell Rep 26:1297–1307
Guthridge KM, Dupal MP, Kölliker R, Jones ES, Smith KF, Forster JW (2001) AFLP analysis of genetic diversity within and between populations of perennial ryegrass (Lolium perenne L.). Euphytica 122:191–201
Hao Y-J, Wen X-P, Deng X-X (2004) Genetic and epigenetic evaluations of citrus calluses recovered from slow-growth culture. J Plant Physiol 161:479–484
Hegarty MJ, Hiscock SJ (2008) Genomic clues to the evolutionary success of polyploid plants. Curr Biol 18:R435–R444
Joyce SM, Cassells AC (2002) Variation in potato microplant morphology in vitro and DNA methylation. Plant Cell Tissue Organ Cult 70:125–137
Kuznetsova OI, Ash OA, Gostimsky SA (2006) The effect of the duration of callus culture on the accumulation of genetic alterations in pea Pisum sativum L. Russ J Genet 42:555–562
Lakshmanan V, Venkataramareddy SR, Neelwarne B (2007) Molecular analysis of genetic stability in long-term micropropagated shoots of banana using RAPD and ISSR markers. Electron J Biotechnol 10:1–8
Laurentin H, Karlovsky P (2007) AFLP fingerprinting of sesame (Sesamum indicum L.) cultivars: identification, genetic relationship and comparison of AFLP informativeness parameters. Genet Resour Crop Evol 54:1437–1446
Li X, Yu X, Wang N, Feng Q, Dong Z, Liu L, Shen J, Liu B (2007) Genetic and epigenetic instabilities induced by tissue culture in wild barley (Hordeum brevisubulatum (Trin.) link). Plant Cell Tissue Organ Cult 90:153–168
Loria R, Bukhalid RA, Fry BA, King RR (1997) Plant pathogenicity in the genus Streptomyces. Plant Dis 81:836–846
Lucht JM, Mauch-Mani B, Steiner H-K, Metraux J-P, Ryals J, Hohn B (2002) Pathogen stress increases somatic recombination frequency in Arabidopsis. Nat Genet 30:311
Mantel NA (1967) The detection of disease clustering and a generalized regression approach. Cancer Res 27:209–220
Matthes M, Singh R, Cheah S-C, Karp A (2001) Variation in oil palm (Elaeis guineessis Jacq.) tissue culture-derived regenerants revealed by AFLPs with methylation-sensitive enzymes. Theor Appl Genet 120:971–979
McClelland M, Nelson M, Raschke E (1994) Effect of site-specific modification on restriction endonucleases and DNA modification methyltransferases. Nucleic Acids Res 22:3640–3659
McGregor CE, Lambert CA, Greyling MM, Louw JH, Warnich L (2000) A comparative assessment of DNA fingerprinting techniques (RAPD, ISSR AFLP and SSR) in tetraploid potato (Solanum tuberosum L.) germplasm. Euphytica 113:135–144
Nielsen SL, Bång H, Kotkas K, Kristensen K, Palohuhta JP, Rosenberg V, Tolstrup K (2007) Variation of growth and disease characters between clones of potato (Solanum tuberosum L.). Potato Res 50:97–114
Pacheco G, Cardoso SRS, Gagliardi RF, Valls JFM, Ferreira PCG, Cardoso MA, Mansur E (2008) Genetic and epigenetic analyses of in vitro-grown plants of Arachis villosulicarpa Hoehne (Leguminosae) obtained from seed explants through different regeneration pathways. J Hortic Sci Biotechnol 83:737–742
Pavet V, Quintero C, Cecchini NM, Rosa AL, Alvarez ME (2006) Arabidopsis displays centromeric DNA hypomethylation and cytological alterations of heterochromatin upon attack by Pseudomonas syringae. Mol Plant Microbe Interact 19:577–587
Peredo EL, Revilla MA, Arroyo-García R (2006) Assessment of genetic and epigenetic variation in hop plants regenerated from sequential subcultures of organogenic calli. J Plant Physiol 163:1071–1079
Phillips RL, Kaeppler SM, Olhoft P (1994) Genetic instability of plant tissue cultures: breakdown of normal controls. Proc Natl Acad Sci 91:5222–5226
Richards EJ (2006) Inherited epigenetic variation-revisiting soft inheritance. Nat Rev Genet 7:395–401
Rodríguez-López CM, Wetten AC, Wilkinson MJ (2010) Progressive erosion of genetic and epigenetic variation in callus-derived cocoa (Theobroma cacao) plants. New Phytol 186:856–868
Saze H (2008) Epigenetic memory transmission through mitosis and meiosis in plants. Semin Cell Develop Biol 19:527–536
Schellenbaum P, Mohler V, Wenzel G, Walter B (2008) Variation in DNA methylation patterns of grapevine somaclones (Vitis vinifera L.). BMC Plant Biol 8:78–88
Sha AH, Lin XH, Huang JB, Zhang DP (2005) Analysis of DNA methylation related to rice adult plant resistance to bacterial blight based on methylation-sensitive AFLP (MSAP) analysis. Mol Gen Genomics 273:484–490
Sharma SK, Bryan GJ, Winfield MO, Millam S (2007) Stability of potato (Solanum tuberosum L.) plants regenerated via somatic embryos, axillary bud proliferated shoots, microtubers and true potato seeds: a comparative phenotypic, cytogenetic and molecular assessment. Planta 226:1449–1458
Stansfield WD (2006) Luther Burbank: honorary member of the American breeders’ association. J Hered 97:95–99
Vazquez AM (2001) Insight into somaclonal variation. Plant Biosyst 135:57–62
Vazquez AM, Linacero R (2010) Stress and somaclonal variation. In: Pua E-C, Davey MR (eds) Plant developmental biology: biotechnological perspectives. Springer, Heidelberg, Germany
Visser RGF, Bachem CWB, de Boer JM, Bryan GJ, Chakrabati SK, Feingold S, Gromadka R, van Ham RCHJ, Huang S, Jacobs JME, Kuznetsov B, de Melo PE, Milbourne D, Orjeda G, Sagredo B, Tang X (2009) Sequencing the potato genome: outline and first results to come from the elucidation of the sequence of the world’s third most important food crop. Am J Potato Res 86:417–429
Vos P, Hogers R, Bleeker M, Reijans M, van de Lee T, Hornes M, Frijters A, Pot J, Peleman J, Kuiper M, Zabeau M (1995) AFLP: a new technique for DNA fingerprinting. Nucleic Acids Res 23:4407–4414
Wada Y, Miyamoto K, Kusano T, Sano H (2004) Association between up-regulation of stress-responsive genes and hypomethylation of genomic DNA in tobacco plants. Mol Gen Genomics 271:658–666
Wilson CR (2004) A summary of common scab disease of potato research from Australia. In: Naito S, Kondo N, Akino S, Ogoshi A, Tanaka F (eds) International potato scab symposium 2004. Hokkaido University, Japan
Wilson CR, Ransom LM, Pemberton BM (1999) The relative importance of seed-borne inoculum to common scab disease of potato and the efficacy of seed tuber and soil treatments for disease control. J Phytopathol 147:13–18
Wilson CR, Tegg RS, Wilson AJ, Luckman GA, Eyles A, Yuan ZQ, Hingston LH, Conner AJ (2010) Stable and extreme resistance to common scab of potato obtained through somatic cell selection. Phytopathology 100:460–467
Xu M, Li X, Korban SS (2000) AFLP-based detection of DNA methylation. Plant Mol Biol Rep 18:361–368
Zilberman D, Henikoff S (2007) Genome-wide analysis of DNA methylation patterns. Development 134:3959–3965
Zilberman D, Coleman-Derr D, Ballinger T, Henikoff S (2008) Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature 456:125–129
Acknowledgments
The authors would like to thank Annabel Wilson for tissue culture and maintenance of all the potato lines, Dr. Robert Tegg for data and experimental information on field and pot trials, Peter Molesworth for help with statistical analysis and Adam Smolenski of the University of Tasmania, Central Science Laboratory for organising samples to be processed on the Beckman Coulter CEQ8000. This project had partial funding from an internal University of Tasmania grant.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by H. Judelson.
Rights and permissions
About this article
Cite this article
Dann, A.L., Wilson, C.R. Comparative assessment of genetic and epigenetic variation among regenerants of potato (Solanum tuberosum) derived from long-term nodal tissue-culture and cell selection. Plant Cell Rep 30, 631–639 (2011). https://doi.org/10.1007/s00299-010-0983-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-010-0983-9