
Abstract
Plants in the family Solanaceae are used as model systems in comparative and evolutionary genomics. The complete chloroplast genomes of seven solanaceous species have been sequenced, including tobacco, potato and tomato, but not peppers. We analyzed the complete chloroplast genome sequence of the hot pepper, Capsicum annuum. The pepper chloroplast genome was 156,781 bp in length, including a pair of inverted repeats (IR) of 25,783 bp. The content and the order of 133 genes in the pepper chloroplast genome were identical to those of other solanaceous plastomes. To characterize pepper plastome sequence, we performed comparative analysis using complete plastome sequences of pepper and seven solanaceous plastomes. Frequency and contents of large indels and tandem repeat sequences and distribution pattern of genome-wide sequence variations were investigated. In addition, a phylogenetic analysis using concatenated alignments of coding sequences was performed to determine evolutionary position of pepper in Solanaceae. Our results revealed two distinct features of pepper plastome compared to other solanaceous plastomes. Firstly, large indels, including insertions on accD and rpl20 gene sequences, were predominantly detected in the pepper plastome compared to other solanaceous plastomes. Secondly, tandem repeat sequences were particularly frequent in the pepper plastome. Taken together, our study represents unique features of evolution of pepper plastome among solanaceous plastomes.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Chloroplasts are organelles presumed to be of endosymbiotic origin from free-living cyanobacteria (Gray 1989). Over one billion years of evolution, eukaryotic chloroplast genes have mostly been transferred to the nucleus (Wilson et al. 1987; Sugiura 1989). As a result, the chloroplast genomes of the majority of extant land plants contain only 90–110 unique genes out of 115–165 kb of DNA (Jansen et al. 2005). The products of these genes constitute about 10–20% of plastid proteins, while nucleus encoded proteins are responsible for the remainder (Sugiura 1989).
The complete chloroplast genomes of more than 170 species including protists, thallophytic, bryophytic and vascular plants have been analyzed. (http://chloroplast.ocean.washington.edu/cpbase/run). Analyses of these sequences show that chloroplast genome structure and gene contents are highly conserved among land plants (Daniell et al. 2006; Leseberg and Duvall 2009). Most chloroplast genomes are quadripartite in structure which includes two single copy DNA fragments, a large single copy (LSC) and a small single copy (SSC), separated by a pair of inverted repeats (IRs) on a single circular molecule (Sugiura et al. 1998). The stabilization effect of these large IRs along with the compact gene arrangement and paucity of dispersed repeats may be responsible for the highly conserved structures of chloroplast genomes (Palmer 1990).
Although overall chloroplast organization is highly conserved among taxa, rearrangements or structural variations have been detected in several chloroplast genomes, which include inversions (Greiner et al. 2008; Kim and Lee 2005), gene duplications (Palmer et al. 1987; Chumley et al. 2006) and loss of IRs (Saski et al. 2005, Wakasugi et al. 1994). Large inversions, resulting in rearrangements of intervening genes, were detected in broad range of taxa including Funariaceae (Goffinet et al. 2007), Fabaceae (Kato et al. 2000), Campanulaceae (Cosner et al. 1997), Geraniaceae (Palmer et al. 1987), Onagraceae (Greiner et al. 2008), Cyatheaceae (Gao et al. 2009) and Poaceae (Doyle et al. 1992; Michelangeli et al. 2003). Recent studies have shown that the end points of inversion sequences usually contain large numbers of short repeats or tRNA sequences, implying an unknown mechanism for this process (Greiner et al. 2008; Haberle et al. 2008). In addition to structural differences, sequence variations have also been investigated by comparative analyses of closely related species. For instance, alignment of the entire chloroplast genome sequences of related cereal species (Bortiri et al. 2008; Leseberg and Duvall 2009) and five Oenothera species (Greiner et al. 2008) showed that a number of sequence polymorphisms were generated during plastome evolution and that highly divergent sequences are concentrated in specific regions called “hotspots”. Sequence polymorphisms have been used to define phylogenetic relationships between species and to understand speciation events associated with cytoplasm–nucleus incompatibility (Greiner et al. 2008).
The family Solanaceae consists of 3,000 species including familiar crops such as tomato, potato, eggplant, petunia, tobacco and pepper. Not only it is the most economically important plant family in horticulture, but it also provides useful model systems for fruit development (tomato and pepper), disease resistance (potato and tomato), synthesis of secondary metabolites such as anthocyanin (petunia) and capsaicin (pepper; Wang et al. 2008). Complete chloroplast genomes are known for seven species: Nicotiana tabacum (Shinozaki et al. 1986), N. sylvestris (Yukawa et al. 2006), N. tomentosiformis (Yukawa et al. 2006), Atropa belladonna (Schmitz-Linneweber et al. 2002), Solanum bulbocastanum (Daniell et al. 2006), S. tuberosum (Chung et al. 2006) and S. lycopersicum (Daniell et al. 2006; Kahlau et al. 2006). Comparative analyses of these sequences shed light on the evolution of plastomes of closely related species among plant families and have shown a significant number of sequence variations including single nucleotide polymorphisms (SNPs), insertions and deletions (indels) in addition to variations in RNA editing patterns between plastomes, although overall plastome structures and gene contents are conserved (Chung et al. 2006; Daniell et al. 2006; Kahlau et al. 2006). These data have been used to investigate the mechanisms of speciation and evolutionary relationships between solanaceous species (Schmitz-Linneweber et al. 2002).
Pepper is a major solanaceous crop that is economically important worldwide. Although an EST database (Kim et al. 2008) and genetic maps have been constructed for pepper, the available sequence information is insufficient to characterize the evolution of pepper among the Solanaceae compared to other crops such as tomato, potato and tobacco. Moreover, reports of organellar genome sequences of peppers are scarce, except for studies in which specific genes in organellar genomes were used for phylogenetic analysis (Bohs and Olmstead 1997; Olmstead et al. 2008).
In this study, we report the complete sequence of the chloroplast genome in the chili pepper, Capsicum annuum. Comparative analyses of eight solanaceous plastomes, including the pepper plastome, were performed to provide information regarding the evolution of the pepper plastome in Solanaceae and overall characteristics of solanaceous chloroplast genomes by detecting sequence variations at the genome-wide level.
Materials and methods
Plant materials
Chili pepper cultivar (C. annuum L.) ‘FS4401’ was provided by Monsanto Korea and used for plastid sequence analysis.
DNA extraction
The mitochondrial fraction was isolated from seedlings grown in a dark room for 3 weeks following the method of Kim et al (2007). Although the majority of the isolated fraction was mitochondrial, a small quantity of plastids was included in the fraction as contamination is inevitable during mitochondrial isolation (Gillman et al. 2007). DNA was extracted from the fraction using DNaseI method (Kim et al. 2007)
DNA sequencing and sequence assembly
Sequences of isolated DNA were analyzed by the GS-FLX pyrosequencing method (Margulies et al. 2005) using the Genome Sequencer FLX system (Roche, Basal, Switzerland). A total of 235,686 sequences, with an average fragment length of 247 bp, were analyzed to generate 58,113,817 bp sequences in total. The sequences were assembled using the CAP3 program (Huang and Madan 1999). Although the major portion of the analyzed DNA was from the mitochondria, extremely high sequence coverage enabled us to assemble contigs covering most of the plastid genome. A total of 12 contigs longer than 2 kb were shown to contain plastid DNA sequences using Basic Local Alignment Search Tool (BLAST; http://blast.ncbi.nlm.nih.gov/) and these contigs covered 137 kb of the plastid genome in total. Gaps between contigs were filled by direct sequencing of PCR product amplified from primers designed using the end sequences of each contig. An additional 17 PCR reactions, amplifying contig regions that were highly divergent from other solanaceous chloroplast genomes, were performed using DNA from green leaves. The sequences of PCR products were directly analyzed to confirm validity of the contig assembly.
Gene annotation, sequence alignment and repeat prediction
Chloroplast genes were annotated on the chloroplast genome sequence of FS4401 using the Dual Organellar GenoMe Annotator (DOGMA; Wyman et al. 2004). This program uses BLASTX against 16 chloroplast genomes of plants to identify chloroplast genes in query sequences.
The complete chloroplast sequences of seven other solanaceous species were obtained from GenBank for comparative plastome analysis: Nicotiana tabacum [Z00044.2], N. sylvestris [AB237912.1], N. tomentosiformis [AB240139.1], Atropa belladonna [AJ316582.1], Solanum bulbocastanum [DQ347958.1], S. tuberosum [DQ386163.1] and S. lycopersicum [AM087200.3]. Pairwise alignments between eight solanaceous plastid genomes were performed using the mVISTA program (Frazer et al. 2004) in Shuffle-LAGAN mode. In this condition, a cut-off of 70% identity is used for the plot and percent identity (50 – 100%) is represented along the Y axis. Tandem repeat sequences were predicted by Tandem repeats finder program (Benson 1999). Alignment parameters were set as 2, 7, 7 for match, mismatch, and indels, respectively. The minimum alignment score and maximum period size were set as 50 and 500, respectively.
Phylogenetic analysis of concatenated chloroplast proteins
We classified 75 chloroplast genes which encode proteins and performed sequence alignment for each gene using bioinformatics tools on Comparative Fungal Genomics Platform (http://cfgp.snu.ac.kr/; Park et al. 2008). Among a total of 79 protein coding genes, accD, rpl20 and ycf1 which show high level of variation or contain large indels and ycf15 which does not exist on the plastome of outgroup species (Coffea arabica and Daucus carota) were excluded in this analysis. Each aligned sequence was curated manually and the sequences were then concatenated. Phylogenetic analysis of seven solanaceous plastomes and two outgroup plastomes was supported by ClustalW 1.83 (Thompson et al. 2002) using the concatenated nucleotide sequences. A maximum likelihood tree was constructed using the PhyML v3.0 (http://www.atgc-montpellier.fr/phyml) with 500 bootstrap iterations.
Results and discussion
Assembly of the C. annuum plastid genome
Plastid genome sequences were obtained as a byproduct of the sequencing of DNA isolated from a fraction of cell organelles of C. annuum. The majority of the C. annuum plastid genome sequence, constituting 137 kb in total, was successfully assembled. Although plastid DNA is often integrated into the mitochondrial genome in plant species (Clifton et al. 2004; Sugiyama et al. 2005), the contig sequences we used in the assembly of the pepper plastome appeared to be originated from plastid rather than mitochondria for two reasons. First, no redundant contigs were obtained for any of the selected contigs. Second, we did not detect any significant mutations resulting in frame shifts or abrupt appearance of stop codons in any of the contig sequences; plastid sequences that are integrated into mitochondrial genomes are usually associated with gene sequences that are non-functional and prone to mutation. Meanwhile, gap sequences between contigs were expected to correspond to plastid genome portions that were integrated into the mitochondrial genome. For instance, a gap was detected on the upstream region of the accD gene during contig assembly, and a sequence with high similarity to this region is also found in the mitochondrial genome (Jo et al. 2009). The redundancy of these sequences might prevent the assembly of contigs containing plastid sequences. The complete plastome sequence was obtained through sequence analysis of PCR products that were amplified by primers designed using end sequences of plastid-specific contigs and shown to contain single sequences by chromatogram analysis.
Organization and gene contents of pepper chloroplast genome
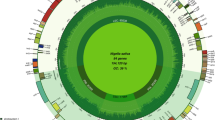
The estimated size of the pepper plastid genome is 156,781 bp, which is the largest among known solanaceous plastomes. The quardripartite structure includes 87,366 bp of LSC and 25,783 bp of SSC that are separated by a pair of 17,849 bp of IR copies (Fig. 1; Table 1). The GC content is 37.7%, which is consistent with other solanaceous plastomes. Coding sequences constitute 58.5% of the pepper plastome sequence. There are 113 unique genes among which 20 are duplicated in IR sequences. A total of 79 unique genes (6 duplicated) encode proteins including photosynthesis-related proteins (46 genes), genetic system related proteins (27 genes), proteins with unique function such as acetyl-CoA carboxylase subunit (accD) and heme attachment to cytochrome C (ccsA) and proteins with unknown functions (ycfs). In addition, 30 unique genes (7 duplicated) and 4 unique duplicated genes encode for tRNAs and rRNAs, respectively. The gene contents and gene order of C. annuum were identical to those of the seven previously known solanaceous plastomes (Table 1).

Gene map of the chloroplast genome of Capsicum annuum L. The genes drawn outside of the circle are transcribed clockwise and inside are transcribed counterclockwise. The colors of the genes are classified according to the functions of the gene products
Pepper-specific gene structure and orfs
Variations in the length of coding sequences in C. annuum are significantly longer than indels usually found in other gene sequences, for example: large insertions were detected on accD and rpl20 sequences in the comparison of pepper gene sequences and have counterparts in other solanaceous plastomes. A 144 bp insertion was detected in the pepper plastome in accD 674 bp downstream of the start codon for this gene (Fig. 2a). Seven repeats of a 18 bp-long motif sequence were observed in the inserted sequence. In addition, pairs of 15 bp-long direct repeat sequences were found in both flanking regions of the inserted sequence. The accD gene encodes the carboxyltransferase β subunit of acetyl-coenzyme A carboxylase (ACCase) which is required for normal growth of dicot plant species. However, the gene sequence is absent from the plastomes of monocot species (Chung et al. 2006; Kode et al. 2005). The accD gene is one of the most variable plastid genes and is probably under diversifying selection (Daniell et al. 2006). The 5′ region of the accD gene was shown to be less conserved than the 3′ region (Lee et al. 2004). Because a pepper-specific insertion was detected on the 5′ region without resulting frame shift, and the trancription of this gene was confirmed by reverse transcriptase PCR (RT-PCR; data not shown), the product of the unusual accD gene of C. annuum is expected to be functional in C. annuum. An insertion on the 3′ end region of rpl20 resulted in the disruption of the stop codon that is normally present in the rpl20 of other solanaceous plastomes (Fig. 2b). Instead, a new stop codon was found 80 bp downstream of the divergent sequence, increasing the length of the coding sequence of the rpl20 gene. Although functional studies of rpl20 have not yet been performed, we expect that rpl20 may be functional in C. annuum because rpl20 is transcribed in C. annuum (data not shown) and 3′ end of this gene is not conserved well in other species (Fig. 2b).

Comparison of pepper DNA and protein sequences of genes that contain large insertions. a Sequence comparison of DNA (upper) and protein (lower) region of accD gene. The repeat sequences at the ends of the inserted DNA fragment are surrounded by yellow-dotted boxes and repeat sequences inside the inserted fragment are enclosed by red boxes. b Comparison of DNA (upper) and protein (lower) sequences of 3′ and C terminus region of the rpl20 gene
In addition to gene sequences, four open reading frames (orfs) were shown to be conserved in length among plastomes of seven solanaceous plastomes (Chung et al. 2006). Two orfs (orf79, orf71B) out of these four orfs were conserved in the C.annuum plastome whereas the other two (orf70B, orf131) were shortened by the creation of early stop codon due to 4 bp insertions. Although two orfs were conserved in length among eight solanaceous plastomes, experimental evidences should be presented to consider these orfs as genes because the length of orfs are short and orfs are located on IR sequences where the mutation rate is much lower than LSC or SSC regions due to frequent gene conversion (Khakhlova and Bock 2006).
Expansions and contractions of IRs
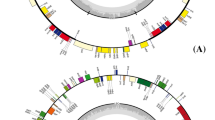
The ends of IRs were expanded or contracted according to the plastomes (Fig. 3). The IRs of A. belladonna contained 1,438 bp of a 5′ portion of the ycf1 gene at one end, while the IRs of N. tabacum included only 996 bp of this region. The other IR end of S. lycopersicum was expanded and included 91 bp of rps19 gene 3′ while that of N. tabacum was contracted and contained none of rps19 coding region. The IRs of pepper consist of 1,094 bp of ycf1 and 65 bp of rps19, showing moderate expansion/contraction of IRs among solanaceous plastomes. Overall, the lengths of IR sequences were not consistent with the total sizes of plastomes.

Contraction and expansion of IR sequences among solanaceous plastomes. The representation for solanaceous plastomes except for pepper plastome was adapted from Chung et al (2006). Plastomes of N. sylvestris and S. bulbocastanum were not represented because contraction and expansion patterns in these species are the same as those of N. tabacum and S. tuberosum, respectively
Variations in large indels
A total of 12 insertions or deletions, of which the largest gap size is over 100 bp, were found by sequence alignment of solanaceous plastomes (Table 2). The pepper (C. annuum) plastome contained the largest number (9 out of 12) of insertions while many were deleted in the S. tuberosum plastome. Among nine sequences inserted in the pepper plastome, two sequences inserted in the trnL–trnF intergenic region and accD gene sequence, respectively, were specific for C. annuum, but other highly similar sequences were not found by BLAST search. Most large insertions were identified in LSC region of C. annuum plastome while the contraction or expansion patterns of IR sequences were similar to other solanaceous plastomes. Meanwhile, specific loss of a 498 bp sequence in the C. annuum plastome may explain why the SSC region of C. annuum plastome is the shortest among solanaceous plastomes.
Some insertions or deletions were genus specific: for example, insertions in trnL–trnF and accD, deletions in petA–psbJ and ndhF–rpl32 for Capsicum, an insertion in trnS–trnG, a deletion in ycf4–ycf10 for Nicotiana and deletions in trnE–trnT, psbE–petL, infA–rps8 for Solanum. A species-specific deletion was also detected in the plastome of S. tuberosum (ndhC–trnV). Most of the inserted or deleted sequences were located in the LSC region, except for two indels which were located in IRs and the SSC region. For many indels, the border positions could not be clearly defined. Instead, intermediate length sequences with different borders were detected rather than complete insertions or deletions. Border positions for these kinds of sequences were largely conserved in each genus.
Among 12 sequences, 9 contained repeated sequences on the borders and all of large indels specific for Solanaceae were included in those sequences. This implies that Solanaceae-specific large indels may be acquired during the divergence of Solanaceae. However, 3 out of 12 sequences were not involved in repeated sequences and none of Solanaceae-specific sequences were included in these sequences. Instead, deletions were detected for the three indels in a single genus or species: deletion of the sequence on ndhC–trnV in S. tuberosum, ycf4–ycf10 in Nicotiana and petA–psbJ in C. annuum. Insertions or deletions of large sequences have been shown to be associated with short direct repeats located at the terminal of the indel sequences in many plant species (Kim and Lee 2004; Milligan et al. 1989; Ogihara et al. 1988; Shimada and Sugiura 1989). Differently from small indels which may be originated by slipped-strand mispairing mechanism (Levinson and Gutman 1987), large indels have been suggested to be generated by illegitimatic recombination events using terminal direct repeat sequences (Kim and Lee 2004). The nine large indels with direct repeats may be originated by this mechanism. Difference in the frequency of recombination events between species might influence the patterns of insertion or deletion in each species. Meanwhile, three indels were not associated with direct repeats. In these cases, other mechanisms besides illegitimatic recombination may be responsible for the acquisition or loss of chloroplast sequences, or extensive sequence modifications may have occurred after insertions or deletions from the illegitimatic recombination.
Variations in tandem repeat sequences
The total length of tandem repeats in the pepper plastome was significantly longer than other solanaceous plastomes including Atropa, Nicotiana and Solanum (Fig. 4a). Proliferation of tandem repeats in certain species has been reported for other families (Greiner et al. 2008; Haberle et al. 2008; Ogihara et al. 1988). For example, the plastome of Trachelium caeruleum contains an extremely high number of tandem repeats compared to other species in the same genus. These tandem repeats were shown to be related to the highly rearranged structure of the T. caeruleum plastome (Haberle et al. 2008). However, no rearrangement was detected near the tandem repeat sequences in the pepper plastome when compared to other solanaceous plastomes, although the pepper plastome contained significantly larger number of tandem repeat sequences than other solanaceous plastomes. A comparison of number of tandem repeats and total length of tandem repeats between species revealed highly similar patterns and ratios indicating that differences in tandem repeat contents among plastomes are mainly comprised of the presence or absence of specific repeats but not the number of copies of a given repeat (Fig. 4). The classification of tandem repeats according to presence or absence of sequences on specific plastomes showed that a high portion of tandem repeats are shared between closely related species included in the same genus (Table 3). However, most of tandem repeat contents of C. annuum and A. belladonna were specific to their plastomes, indicating that those tandem repeat sequences were generated after divergence of C. annuum or A. belladonna from other solanaceous species. This result was different from previous research where direct or inverted repeats were shown to be highly conserved between solanaceous plastomes (Daniell et al. 2006). This inconsistency may be due to differences in definitions of repeats and the fact that conservation of repeats was determined only for repeats detected in the tobacco plastome by previous research. Although tandem repeat contents are not well-conserved between genera, the pattern of distribution of tandem repeats was highly consistent between plastomes (Fig. 5). This implies that the processes that generate new tandem repeats have occurred in specific locations of the plastid sequence. The positions of regions in which large amount of tandem repeats were contained were different from those of regions where sequence divergence between species is high, indicating that uncorrelation with plastid gene function is not a sufficient requirement for sequences prone to generate tandem repeats.

Comparison of tandem repeat contents between Solanaceous plastomes and two other plastomes. a Total length of tandem repeats. b Total number of tandem repeats

Distribution of tandem repeats on solanaceous plastomes. Total lengths of tandem repeat sequences were surveyed on every 1 kb of plastome sequences
Although unequal recombination and slipped-strand mispairing during DNA replication have been suggested as possible mechanisms for the generation of tandem repeats (Levinson and Gutman 1987), it is unclear which factors are responsible for the large content of tandem repeat on pepper plastome. The fact that majority of pepper tandem repeats were detected specifically on pepper plastome implies that the factors which increased the instability of plastome has been evolved after pepper was diverged from other solanaceous species. Further analysis of the complete plastome sequences of more closely related taxa and the comparison of the recombination or replication patterns in chloroplast of solanaceous species may suggest the evolutionary process by which the noble characteristics of pepper plastome were acquired.
Pair-wise alignments of solanaceous plastomes
Pair-wise alignments of solanaceous plastomes were performed for genome-wide comparison. Although the degree of variation differed between alignment combinations, the distribution patterns of sequence variation were highly consistent between alignment combinations (Fig. 6). With the exception of ycf1which is known to reflect diversifying selection (Greiner et al. 2008), sequence variations were detected mainly in intergenic or intron regions. Intergenic regions with high degrees of divergence included rps16–trnQ, rbcL–accD, trnE–trnG and trnT–trnfM, in which DNA rearrangements were detected in Oenothera or fern species (Gao et al. 2009). Also, tRNA gene clusters and the regions between rbcL and cemA have been reported as hot spots in the glass family (Calsa Junior et al. 2004; Maier et al. 1995; Ogihara et al. 2002) were included. In the SSC, the region between ndhF and trnL was highly variable between species and also contained large deletions in pepper. When the degrees of sequence divergence in LSC, SSC and IR regions were compared, IR regions were showed to be conserved compared to other regions, possibly due to copy correction between IR sequences by gene conversion (Khakhlova and Bock 2006). Sites where large insertions or deletions (>100 bp) occurred were mainly located on highly variable sequences on LSC or SSC regions, indicating that those variable sequences are not requisite for normal function of plastomes.

Pair-wise comparison of plastome sequences between four species in Solanaceae using the VISTA program. The Y scale represents % identity ranging from 50–100%. The blue regions are exons and red regions are non-coding. Indel sequences longer than 100 bp are represented by red arrows at the bottoms of plastome sequence alignments that contain the longest insertion sequence for each respective indel. IR regions for the pepper plastome are represented on the pepper plastome alignments
Phylogenetic analysis of solanaceous plastomes
Several chloroplast genes such as matK and rbcL or intergenic sequences trnH–psbA and trnL–trnF have been used for phylogenetic analysis between diverse plant species due to sequence conservation among plant taxa in tandem with suitable variation to deduce the evolutionary relationships between species (Lahaye et al. 2008; Taberlet et al. 1991, 2007). However, determination of phylogeny depending on single gene sequences may be inaccurate (Guo et al. 2007). The use of many individual chloroplast genes or concatenated gene sequences in phylogenetic analysis is feasible since complete chloroplast genome sequences are available in many species (Guo et al. 2007; Jansen et al. 2006; Moore et al. 2010).
Although plastome-scale phylogenetic analyses were performed for solanaceae species in which complete plastid sequences were analyzed (Jansen et al. 2007; Moore et al. 2010), the evolutionary position of Capsicum in Solanaceae has been determined only by a single or a few plastid genes (Bohs and Olmstead 1997; Olmstead et al. 2008), a non-coding region on plastid genome (Melotto-Passarin et al. 2008), and specific nuclear DNA regions (Wang et al. 2008). To obtain reasonable phylogenetic relationships using plastome sequence information, we performed multiple sequence alignments for each protein coding gene in a variety of solanaceous plastomes. After concatenating each alignment, maximum likelihood phylogenetic tree was drawn (Fig. 7). In this analysis, taxa were divided into two clades with 100% bootstrap values. The first clade included species in Solanum, Capsicum and Atropa. In this clade, Atropa was separated from the sister taxon of a clade containing Capsicum and Solanum. The second clade consisted of three Nicotiana species. This result was consistent with a previous phylogenetic analysis of plastid ndhF and trnL–F sequences (Olmstead et al. 2008), but different from an analysis of 13 orfs of solanaceous plastomes in which Atropa and Nicotiana were grouped together and separated from Solanum (Chung et al. 2006).

Maximum likelihood phylogram derived using concatenated nucleotide sequences of 75 protein-coding genes of seven solanaceous species and two outgroup species
In this study, we firstly reported the complete sequence of pepper plastid DNA. The plastome was characterized by comparative analysis with other solanaceous plastomes and the evolutionary position of pepper in Solanaceae was determined by phylogenetic analysis using concatenated alignments of gene sequences. Comparison of distribution patterns of large indels and tandem repeats showed that pepper plastome has uniquely frequent large indel and tandem repeat sequences compared to other solanaceous plastomes although other attributes such as gene contents and order, the distribution pattern of variations on plastome were highly conserved. We expect that further detailed analysis using plastomes of a larger number of solanaceous species may enable to deduce the evolutionary process by which pepper plastome acquire these noble characteristics.
References
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27:573–580
Bohs L, Olmstead RG (1997) Phylogenetic relationships in Solanum (Solanaceae) based on ndhF sequences. Syst Bot 22:5–17
Bortiri E, Coleman-Derr D, Lazo GR, Anderson OD, Gu YQ (2008) The complete chloroplast genome sequence of Brachypodium distachyon: sequence comparison and phylogenetic analysis of eight grass plastomes. BMC Res Notes 1:61
Calsa Junior T, Carraro DM, Benatti MR, Barbosa AC, Kitajima JP, Carrer H (2004) Structural features and transcript-editing analysis of sugarcane (Saccharum officinarum L.) chloroplast genome. Curr Genet 46:366–373
Chumley TW, Palmer JD, Mower JP, Fourcade HM, Calie PJ, Boore JL, Jansen RK (2006) The complete chloroplast genome sequence of Pelargonium × hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol Biol Evol 23:2175–2190
Chung HJ, Jung JD, Park HW, Kim JH, Cha HW, Min SR, Jeong WJ, Liu JR (2006) The complete chloroplast genome sequences of Solanum tuberosum and comparative analysis with Solanaceae species identified the presence of a 241-bp deletion in cultivated potato chloroplast DNA sequence. Plant Cell Rep 25:1369–1379
Clifton SW, Minx P, Fauron CM, Gibson M, Allen JO, Sun H, Thompson M, Barbazuk WB, Kanuganti S, Tayloe C, Meyer L, Wilson RK, Newton KJ (2004) Sequence and comparative analysis of the maize NB mitochondrial genome. Plant Physiol 136:3486–3503
Cosner ME, Jansen RK, Palmer JD, Downie SR (1997) The highly rearranged chloroplast genome of Trachelium caeruleum (Campanulaceae): multiple inversions, inverted repeat expansion and contraction, transposition, insertions/deletions, and several repeat families. Curr Genet 31:419–429
Daniell H, Lee SB, Grevich J, Saski C, Quesada-Vargas T, Guda C, Tomkins J, Jansen RK (2006) Complete chloroplast genome sequences of Solanum bulbocastanum, Solanum lycopersicum and comparative analyses with other Solanaceae genomes. Theor Appl Genet 112:1503–1518
Doyle JJ, Davis JI, Soreng RJ, Garvin D, Anderson MJ (1992) Chloroplast DNA inversions and the origin of the grass family (Poaceae). Proc Natl Acad Sci USA 89:7722–7726
Frazer KA, Pachter L, Poliakov A, Rubin EM, Dubchak I (2004) VISTA: computational tools for comparative genomics. Nucleic Acids Res 32:W273–W279
Gao L, Yi X, Yang YX, Su YJ, Wang T (2009) Complete chloroplast genome sequence of a tree fern Alsophila spinulosa: insights into evolutionary changes in fern chloroplast genomes. BMC Evol Biol 9:130
Gillman JD, Bentolila S, Hanson MR (2007) The petunia restorer of fertility protein is part of a large mitochondrial complex that interacts with transcripts of the CMS-associated locus. Plant J 49:217–227
Goffinet B, Wickett NJ, Werner O, Ros RM, Shaw AJ, Cox CJ (2007) Distribution and phylogenetic significance of the 71-kb inversion in the plastid genome in Funariidae (Bryophyta). Ann Bot 99:747–753
Gray MW (1989) The evolutionary origins of organelles. Trends Genet 5:294–299
Greiner S, Wang X, Rauwolf U, Silber MV, Mayer K, Meurer J, Haberer G, Herrmann RG (2008) The complete nucleotide sequences of the five genetically distinct plastid genomes of Oenothera, subsection Oenothera: I. sequence evaluation and plastome evolution. Nucleic Acids Res 36:2366–2378
Guo X, Castillo-Ramirez S, Gonzalez V, Bustos P, Fernandez-Vazquez JL, Santamaria RI, Arellano J, Cevallos MA, Davila G (2007) Rapid evolutionary change of common bean (Phaseolus vulgaris L) plastome, and the genomic diversification of legume chloroplasts. BMC Genomics 8:228
Haberle RC, Fourcade HM, Boore JL, Jansen RK (2008) Extensive rearrangements in the chloroplast genome of Trachelium caeruleum are associated with repeats and tRNA genes. J Mol Evol 66:350–361
Huang X, Madan A (1999) CAP3: A DNA sequence assembly program. Genome Res 9:868–877
Jansen RK, Raubeson LA, Boore JL, dePamphilis CW, Chumley TW, Haberle RC, Wyman SK, Alverson AJ, Peery R, Herman SJ, Fourcade HM, Kuehl JV, McNeal JR, Leebens-Mack J, Cui L (2005) Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol 395:348–384
Jansen RK, Kaittanis C, Saski C, Lee SB, Tomkins J, Alverson AJ, Daniell H (2006) Phylogenetic analyses of Vitis (Vitaceae) based on complete chloroplast genome sequences: effects of taxon sampling and phylogenetic methods on resolving relationships among rosids. BMC Evol Biol 6:32
Jansen RK, Cai Z, Raubeson LA, Daniell H, Depamphilis CW, Leebens-Mack J, Muller KF, Guisinger-Bellian M, Haberle RC, Hansen AK, Chumley TW, Lee SB, Peery R, McNeal JR, Kuehl JV, Boore JL (2007) Analysis of 81 genes from 64 plastid genomes resolves relationships in angiosperms and identifies genome-scale evolutionary patterns. Proc Natl Acad Sci USA 104:19369–19374
Jo YD, Jeong HJ, Kang BC (2009) Development of a CMS-specific marker based on chloroplast-derived mitochondrial sequence in pepper. Plant Biotechnol Rep 3:309–315
Kahlau S, Aspinall S, Gray JC, Bock R (2006) Sequence of the tomato chloroplast DNA and evolutionary comparison of Solanaceous plastid genomes. J Mol Evol 63:194–207
Kato T, Kaneko T, Sato S, Nakamura Y, Tabata S (2000) Complete structure of the chloroplast genome of a legume, Lotus japonicus. DNA Res 7:323–330
Khakhlova O, Bock R (2006) Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J 46:85–94
Kim KJ, Lee HL (2004) Complete chloroplast genome sequences from Korean Ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res 11:247–261
Kim KJ, Lee HL (2005) Widespread occurrence of small inversions in the chloroplast genomes of land plants. Mol Cells 19:104–113
Kim DH, Kang JG, Kim BD (2007) Isolation and characterization of the cytoplasmic male sterility-associated orf456 gene of chili pepper (Capsicum annuum L.). Plant Mol Biol 63:519–532
Kim HJ, Baek KH, Lee SW, Kim J, Lee BW, Cho HS, Kim WT, Choi D, Hur CG (2008) Pepper EST database: comprehensive in silico tool for analyzing the chili pepper (Capsicum annuum) transcriptome. BMC Plant Biol 8:101
Kode V, Mudd EA, Iamtham S, Day A (2005) The tobacco plastid accD gene is essential and is required for leaf development. Plant J 44:237–244
Lahaye R, van der Bank M, Bogarin D, Warner J, Pupulin F, Gigot G, Maurin O, Duthoit S, Barraclough TG, Savolainen V (2008) DNA barcoding the floras of biodiversity hotspots. Proc Natl Acad Sci USA 105:2923–2928
Lee SS, Jeong WJ, Bae JM, Bang JW, Liu JR, Harn CH (2004) Characterization of the plastid-encoded carboxyltransferase subunit (accD) gene of potato. Mol Cells 17:422–429
Leseberg CH, Duvall MR (2009) The complete chloroplast genome of Coix lacryma-jobi and a comparative molecular evolutionary analysis of plastomes in cereals. J Mol Evol 69:311–318
Levinson G, Gutman GA (1987) Slipped-strand mispairing: a major mechanism for DNA sequence evolution. Mol Biol Evol 4:203–221
Maier RM, Neckermann K, Igloi GL, Kossel H (1995) Complete sequence of the maize chloroplast genome: gene content, hotspots of divergence and fine tuning of genetic information by transcript editing. J Mol Biol 251:614–628
Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM (2005) Genome sequencing in microfabricated high-density picolitre reactors. Nature 437:376–380
Melotto-Passarin DM, Berger IJ, Dressano K, De Martin VF, Oliveira GCX, Bock R, Carrer H (2008) Phylogenetic relationships in Solanaceae and related species based on cpDNA sequence from plastid trnE–trnT region. Crop Breed Appl Biotechnol 8:85–95
Michelangeli FA, Davis JI, Stevenson DW (2003) Phylogenetic relationships among Poaceae and related families as inferred from morphology, inversions in the plastid genome, and sequence data from the mitochondrial and plastid genomes. Am J Bot 90:93–106
Milligan BG, Hampton JN, Palmer JD (1989) Dispersed repeats and structural reorganization in subclover chloroplast DNA. Mol Biol Evol 6:355–368
Moore MJ, Soltis PS, Bell CD, Burleigh JG, Soltis DE (2010) Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc Natl Acad Sci USA 107:4623–4628
Ogihara Y, Terachi T, Sasakuma T (1988) Intramolecular recombination of chloroplast genome mediated by short direct-repeat sequences in wheat species. Proc Natl Acad Sci USA 85:8573–8577
Ogihara Y, Isono K, Kojima T, Endo A, Hanaoka M, Shiina T, Terachi T, Utsugi S, Murata M, Mori N et al (2002) Structural features of a wheat plastome as revealed by complete sequencing of chloroplast DNA. Mol Genet Genomics 266:740–746
Olmstead RG, Bohs L, Migid HA, Santiago-Valentin E, Garcia VF, Collier SM (2008) A molecular phylogeny of the Solanaceae. Taxon 57:1159–1181
Palmer JD (1985) Comparative organization of chloroplast genomes. Annu Rev Genet 19:325–354
Palmer JD (1990) Contrasting modes and tempos of genome evolution in land plant organelles. Trends Genet 6:115–120
Palmer JD, Nugent JM, Herbon LA (1987) Unusual structure of geranium chloroplast DNA: a triple-sized inverted repeat, extensive gene duplications, multiple inversions, and two repeat families. Proc Natl Acad Sci USA 84:769–773
Park J, Park B, Jung K, Jang S, Yu K, Choi J, Kong S, Park J, Kim S, Kim H, Kim S, Kim JF, Blair JE, Lee K, Kang S, Lee YH (2008) CFGP: a web-based, comparative fungal genomics platform. Nucleic Acids Res 36:D562–D571
Saski C, Lee SB, Daniell H, Wood TC, Tomkins J, Kim HG, Jansen RK (2005) Complete chloroplast genome sequence of Glycine max and comparative analyses with other legume genomes. Plant Mol Biol 59:309–322
Schmitz-Linneweber C, Regel R, Du TG, Hupfer H, Herrmann RG, Maier RM (2002) The plastid chromosome of Atropa belladonna and its comparison with that of Nicotiana tabacum: the role of RNA editing in generating divergence in the process of plant speciation. Mol Biol Evol 19:1602–1612
Shimada H, Sugiura M (1989) Pseudogenes and short repeated sequences in the rice chloroplast genome. Curr Genet 16:293–301
Shinozaki K, Ohme M, Tanaka M, Wakasugi T, Hayashida N, Matsubayashi T, Zaita N, Chunwongse J, Obokata J, Yamaguchi-Shinozaki K, Ohto C, Torazawa K, Meng BY, Sugita M, Deno H, Kamogashira T, Yamada K, Kusuda J, Takaiwa F, Kato A, Tohdoh N, Shimada H, Sugiura M (1986) The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J 5:2043–2049
Sugiura M (1989) The chloroplast chromosomes in land plants. Annu Rev Cell Biol 5:51–70
Sugiura M, Hirose T, Sugita M (1998) Evolution and mechanism of translation in chloroplasts. Annu Rev Genet 32:437–459
Sugiyama Y, Watase Y, Nagase M, Makita N, Yagura S, Hirai A, Sugiura M (2005) The complete nucleotide sequence and multipartite organization of the tobacco mitochondrial genome: comparative analysis of mitochondrial genomes in higher plants. Mol Genet Genomics 272:603–615
Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109
Taberlet P, Coissac E, Pompanon F, Gielly L, Miquel C, Valentini A, Vermat T, Corthier G, Brochmann C, Willerslev E (2007) Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res 35:e14
Thompson JD, Gibson TJ, Higgins DG (2002) Multiple sequence alignment using ClustalW and ClustalX. In: Current protocols in bioinformatics, chap 2, unit 2.3
Wakasugi T, Tsudzuki J, Ito S, Nakashima K, Tsudzuki T, Sugiura M (1994) Loss of all ndh genes as determined by sequencing the entire chloroplast genome of the black pine Pinus thunbergii. Proc Natl Acad Sci USA 91:9794–9798
Wang Y, Diehl A, Wu F, Vrebalov J, Giovannoni J, Siepel A, Tanksley SD (2008) Sequencing and comparative analysis of a conserved syntenic segment in the Solanaceae. Genetics 180:391–408
Wilson AC, Ochman H, Prager EM (1987) Molecular time scale for evolution. Trends Genet 3:241–247
Wyman SK, Jansen RK, Boore JL (2004) Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20:3252–3255
Yukawa M, Tsudzuki T, Sugiura M (2006) The chloroplast genome of Nicotiana sylvestris and Nicotiana tomentosiformis: complete sequencing confirms that the Nicotiana sylvestris progenitor is the maternal genome donor of Nicotiana tabacum. Mol Genet Genomics 275:367–373
Acknowledgments
This work was supported by a grant (code20070301034022) from the BioGreen21 program, by the Rural Development Administration and by the ARC program supported by Agriculture and Forestry, Ministry for Food, Agriculture, Forestry and Fisheries, the Republic of Korea. We thank to Kim, Suk-Jun who kindly helped us with drawing a gene map of pepper chloroplast genome.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Jordan.
Rights and permissions
About this article
Cite this article
Jo, Y.D., Park, J., Kim, J. et al. Complete sequencing and comparative analyses of the pepper (Capsicum annuum L.) plastome revealed high frequency of tandem repeats and large insertion/deletions on pepper plastome. Plant Cell Rep 30, 217–229 (2011). https://doi.org/10.1007/s00299-010-0929-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-010-0929-2