
Abstract
As one of the key tropical crops, coconut (Cocos nucifera L.) is a member of the monocotyledonous family Aracaceae (Palmaceae). In this study, we amplified the upstream region of an endosperm-specific expression gene, Lysophosphatidyl acyltransferase (LPAAT), from the coconut genomic DNA by chromosome walking. In this sequence, we found several types of promoter-related elements including TATA-box, CAAT-box and Skn1-motif. In order to further examine its function, three different 5′-deletion fragments were inserted into pBI101.3, a plant expression vector harboring the LPAAT upstream sequence, leading to pBI101.3-L1, pBI101.3-L2 and pBI101.3-L3, respectively. We obtained transgenic plants of rice by Agrobacterium-mediated callus transformation and plant regeneration and detected the expression of gus gene by histochemical staining and fluorometric determination. We found that gus gene driven by the three deletion fragments was specifically expressed in the endosperm of rice seeds, but not in the empty vector of pBI101.3 and other tissues. The highest expression level of GUS was at 15 DAF in pBI101.3-L3 and pBI101.3-L2 transgenic lines, while the same level was detected at 10 DAF in pBI101.3-L1. The expression driven by the whole fragment was up to 1.76- and 2.8-fold higher than those driven by the −817 bp and −453 bp upstream fragments, and 10.7-fold higher than that driven by the vector without the promoter. Taken together, our results strongly suggest that these promoter fragments from coconut have a significant potential in genetically improving endosperm in main crops.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Endosperm is the storage organ of starch and proteins in cereal crops. It represents 60% of the food supply of the world and provides the major source of calories and proteins for humans. Improving endosperm composition and quality via genetic modification has attracted much attention, and significant progress has been made in this aspect (Bajaj and Mohanty 2005). Compared with other expression systems, endosperm has many advantages as a heterogeneous expression platform, such as cost saving, ease of controlling the production scale, large storage ability, and high safety due to recombinant protein storage and absence of animal-derived pathogens (Takaiwa et al. 2007; Wu et al. 2007). However, only a few strong endosperm-specific expression promoters are known to drive recombinant protein expression in cereal endosperm, which presents a major limitation in improving transgenic mediated crops in the last decade (Furtado et al. 2008). Therefore, it is highly valuable to identify and characterize new endosperm-specific promoters in cereal crops. Foreign protein genes driven by an endosperm-specific promoter are more efficient than those driven by constitutive promoters (Choi et al. 2003).
In genetic transformation, the repetitive use of the same promoter is one major reason for transgenic silencing (Bhullar et al. 2003; Charrier et al. 2000). To achieve the multiple purposes of gene expression in plants, there is a great need for promoters with high and regulated expression of transgenes in monocot seeds (Hiroi and Takaiwa 2006; Furtado et al. 2008). In previous studies, several endosperm-specific expression promoters have been isolated and characterized from rice (Oryza sativa L.) (Rasmussen and Donaldson 2006; Qu and Takaiwa 2004), wheat (Triticum aestivum L.) (Lamacchia et al. 2001; Wiley et al. 2007), maize (Zea mays L.) (Russell and Fromm 1997) and barley (Hordeum vulgare L.) (Choi et al. 2003). However, few promoters with endosperm-specific expression have been available in plant genetic transformation research because of unsatisfied expression level and pattern (Li et al. 2008; Qu et al. 2008).
As one of the key tropical crops, coconut (Cocos nucifera L.) is a member of the monocotyledonous family Aracaceae (Palmaceae). It is the only species of the genus Cocos and belongs to the subfamily Cocoideae that includes 27 genera and 600 species (Rivera et al. 1999, Daniel et al. 2005). The food and industrial products of coconut (such as coconut oil, copra and desiccated coconut) play an important role in the economy of many developing countries. As a special plant in tropics, few genes in coconut have been cloned and characterized. Thus, a large number of unidentified coconut genes provide a great resource for exploring new gene and function (Li and Fan 2008).
LPAAT (1-acyl-sn-glycerol-3-P acyltransferase) is an enzyme, which catalyzes acylation of sn-2 position of Lysophosphatidic acid (LPA) to yield phosphatidic acid (PA) in the Kennedy pathway (Kim and Huang 2004). The PA is dephosphorylated to produce diacylglycerol. Diacylglycerol acyltransferase catalyzes the transfer of a third acyl group to the sn-3 position of the glycerol backbone, which can produce triacylglycerol (TGA) (Okazaki et al. 2006). The acyltransferases, especially LPAAT, have been shown to have a nonrandom distribution (Coleman and Lee 2004; Nath et al. 2009), and they are specifically expressed in the endosperm of coconut. The coding region of LPAAT has been cloned and sequenced from coconut (Knutzon et al. 1995), but the promoter of this gene has not been described in detail. As a gene specifically expressed in the endosperm, the upstream sequence of LPAAT may help us to find a new endosperm-specific promoter.
In this study, we cloned and sequenced the LPAAT promoter from coconut. Then, with an Agrobacterium-mediated transformation of embryogenic callus and plant regeneration, we investigated its function by examining the stable expression of gus gene in the endosperm of rice. Our results provide a novel promoter for producing valuable recombinant proteins and multigene transformation in rice and other cereal crops.
Materials and methods
Materials
Coconut (Cocos nucifera L) leaves were obtained from Institute of Coconut, Chinese Agricultural Academy of Tropical Crops (Wenchang, Hainan, China). Escherichia coli strain DH5α was used for bacterial cloning. The strains were grown at 37°C in LB medium supplemented with kanamycin (50 mg/L) or ampicillin (50 mg/L) according to the plasmid type. Agrobacterium tumefaciens strain EHA105 and plant expression vector pBI101.3 were kindly provided by Professor Lin YJ (National Key Laboratory of Crop Genetic Improvement, Huazhong Agricultural University, Wuhan, China). All chemicals, restriction endonucleases, and other required enzymes were obtained from Sigma (China) or Sangon Biological Engineering T&S Company (Shanghai), unless stated otherwise.
Genomic DNA isolation and upstream region isolation of the LPAAT gene
Coconut high-molecular-weight DNA was isolated from young leaves by the improved CTAB (Cetyltriethylammonium Bromide) method (Liu et al. 2008). The upstream region of LPAAT gene was amplified according to the user manual of Universal Genome Walker Kit (Clontech, Palo Alto, CA, USA). Genomic DNA of coconut was digested with four blunt-end restriction endonucleases (EcoRV, PvuII, DraI and StuI), respectively. The digested products were purified and then ligated to GenomeWalker adaptors. The ligated products were used as template for the first-round nest PCR. The primers (CEP-1: ttcggttgggacatcccgaagctcgctt, and AP1: gtaatacgactcactatagggc) were designed according to the related adaptor and LPAAT sequence. Briefly, the primary PCR amplification was carried out by a two-step cycle: seven cycles at 94°C for 25 s, 72°C for 3 min; 32 cycles at 94°C for 25 s, 67°C for 3 min, and an additional 7 min at 67°C after the final cycle. The primary PCR products were used as template in the secondary PCR amplification with the same program, and the nested primers (CEP-2: gaacttgcccctgaagcatccataggac, and AP2: actatagggcacgcgtggt) were used to detect CEP-1 and AP1, respectively. The product was recovered and ligated to linear vector pMD18-T (TaKaRa, Dalin, China). The sequence analysis was carried out by TaKaRa Biotechnology Company (Dalian, China).
Sequence analysis
In order to predict the function of the upstream region of LPAAT gene, the sequence was analyzed by PlantCARE, a database of plant cis-acting regulatory elements, enhancers, and repressors (http://bioinformatics.psb.ugent.be/webtools/plantcare/html/). Based on the motif location, different 5′-deletion fragments were selected and amplified for expression vector construction.
Plant expression vector construction and promoter deletion analysis
To construct plant expression vectors harboring LPAAT upstream region, three different 5′-deletions were inserted into pBI101.3, leading to pBI101.3-L1, pBI101.3-L2 and pBI101.3-L3, respectively. In order to amplify 5′ LPAAT upstream and its deletions, we synthesized 18–20 bp upstream primers corresponding to the sequence beginning at −453, −817, and −896. The 5′ sequence of each primer was extended 8 bp to generate a HindIII site (underlined). These related primer sequences were as follows: p101-3L: 5′-ataagcttggcacgcgtggtcgacgg-3′, p101-2L: 5′-ataagcttcctgatctattacggtga-3′, p101-1L: 5′-ataagcttccttctttcttgaccgac-3′. A BamHI site (underlined) was added to the 5′ end of the reverse primers 5′-atggatccaggactatatatcccccacttccc-3′. Three amplified products were digested with HindIII and BamHI, and then they were ligated into the likewise digested vector pBI101.3. The related molecular procedures, such as fragment purification, ligation and transformation, were performed as described previously (Sambrook et al. 1989). The pBI101.3 was used as a promoterless control. Plasmid construction and manipulation were carried out according to the previously described standard methods. The constructed vectors were verified by PCR and sequencing (Promoter 101a: 5′-ggacactttatgcttccggctc-3′, and Promoter 101b: 5′-attccacagttttcgcgatcc-3′).
Plant transformation
The obtained four expression vectors (pBI101.3-L3, pBI101.3-L2, pBI101.3-L1 and pBI101.3-L0) were sequenced to verify the gene orientation and transferred into Agrobacterium tumefaciens EHA105 by a freeze–thaw method (Huang et al. 2001). Then, all the constructs were introduced into Zhonghua11 (Oryza sativa L ssp. japonica) by Agrobacterium-mediated transformation. The callus culture and transformation procedures were carried out as in Hiei et al. (1994). Scutellum-derived embryonic calli were co-cultured with the Agrobacterium strain EHA105 with the expression vector for 3 days. Then the calli were transferred to the selection medium containing 50 mg/L G418 and 400 mg/L Carbenicillin. After 8–9 weeks, survived calli on the selection medium were transferred to the pre-regeneration medium containing 50 mg/L G418 and 200 mg/L Carbenicillin. After 7 days, the resistant calli were transferred to the regeneration medium without antibiotic, to regenerate transformed plantlets. The original roots were cut off, and the plantlets were transferred to the rooting medium. After 7 days, the root was washed, and the plantlets were transferred with the vigorous root in the greenhouse.
PCR analysis and Southern blot analysis
PCR and Southern blot analyses were used to determine the presence of gus gene and the copy number in transformed plants. PCR analysis was performed with the primers gus-F (5′-gggcgaacagttcctgatta-3′) and gus-R (5′-aacgtatccacgccgtattc-3′). A 20 μl mixture of 30 ng of template DNA, 1× buffer (50 mM KCl, 10 mM Tris–HCl and 0.1% Triton X-100), 0.15 mM dNTPs, 1.875 mM MgCl2, 0.1 μM of each primer and 1 U of Taq DNA polymerase was prepared for the PCR assay. Initially, the PCR reaction was performed at 94°C for 5 min, then the amplification was carried out with 30 cycles at a melting temperature of 94°C for 1 min, an annealing temperature of 57°C for 1 min, and an extension temperature of 72°C for 1 min, followed by an additional extension at 72°C for 5 min. The PCR products were then analyzed by gel electrophoresis.
For Southern blot analysis, 5 μg of genomic DNA from each sample was digested with HindIII and separated on a 0.8% agarose gel. Subsequently, the purified PCR product was transferred onto a nylon membrane. The probe was prepared from a PCR-amplified gus fragment. All procedures for the hybridization were performed as described previously (Lin and Zhang 2005).
Histochemical β-glucuronidase (GUS) assays
Histochemical localization of GUS activity in transgenic plants was carried out essentially as described previously (Jefferson et al. 1987). In 2008, T0 transgenic plant (transgenic-positive transformants) tissue was incubated in 50 mM sodium phosphate pH 7.0, 0.5 mM potassium ferricyanide, 0.5 mM potassium ferrocyanide, 10 mM Na2-EDTA, 0.1% w/v Triton X-100 and 1 mg/mL 5-bromo-4-chloro-3-indolyl-β-d-glucuronide (X-Gluc) at 37°C for 4–7 h. After staining, the samples were fixed in 70% ethanol, and photographs were taken under a dissecting microscope. More than 10 seeds from each transgenic plant were analyzed. For each construct, five families with high expression level of GUS were selected based on staining levels.
Quantitative GUS activity
In 2009, the selected T1 transgenic plant families were grown on the experimental farm of the Huazhong Agricultural University in Wuhan, China. For each construct, developing endosperm tissues used in fluorometric determination were obtained at 10, 15 and 20 DAF (days after flowering), respectively. Then, the endosperm (at the same developmental stage, but from different families) was mixed as one sample. The samples were grinded in a mortar with liquid nitrogen. Then, 1 mL extraction buffer (0.05 M NaH2PO4 at pH 7.0, 0.01 M EDTA at pH 8.0, 0.1% SDS, 0.1% Triton X-100, and 0.01 M β-mercaptoethanol) was added to ~100 mg of frozen tissue powder, and homogenized by vortexer. The homogenate was centrifuged at 13,000 rpm at 4°C for 10 min, and the supernatant was used for protein quantification and fluorometric assay (Bradford 1976). The fluorometric analysis of GUS activity was performed according to the method described by Jefferson et al. (1987). The substrate 4-methylumbelliferyl β-d-glucuronide (MUG) was added to the supernatant, and then the mixture was incubated at 37°C for 20 min. GUS activity was analyzed in a Microfluor fluorometer (Hitachi) with an emission at 455 nm and an excitation at 365 nm. The fluorometer was calibrated with freshly prepared MU standards in the same buffer prior to the assay. GUS enzyme activity was expressed as pmol of 4-methylumbelliferone (MU) produced per minute per milligram of protein.
Results
Isolation and sequence analysis of the LPAAT up-stream fragment
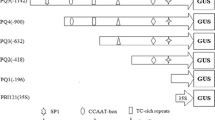
We amplified a 1.2 kb DNA fragment that contains the upstream sequence of the LPAAT gene from coconut genomic DNA by the two-cycle PCR. Then we identified the product with agarose electrophoresis and sequenced the fragment. The sequence analysis showed that a 96 bp sequence of this clone was identical to the LPAAT cDNA sequence, confirming that the correct fragment of upstream sequence of LPAAT was amplified and cloned. As shown in Fig. 1, a 910 bp upstream sequence from the ATG start codon was assigned as the candidate LPAAT promoter region, and it was submitted to PlantCARE to detect putative endosperm-specific elements involved in the regulation of gene expression. We found several types of motifs in this promoter: one putative TATA box (TAATA) (−11), four CAAT boxes (CACAAT and CAAT) (−872, −838, −352 and +18) and four 5′UTR Py-rich stretches (TTTCTTCTCT) (−170, −216, −276 and −326). Moreover, the active role of the CAAT boxes was reinforced by the absence of the GC box. In addition, we also detected one cis-acting regulatory element, Skn-1 motif (−784), which is required for endosperm expression.

Sequence analysis of the upstream region of the LPAAT. The code region of LPAAT gene was underlined. Different types of elements related to endosperm-specific promoter are shown in different kinds of lines. The deletion site of promoter is marker by arrows
Construction of expression vectors for function analysis (gus chimeric genes)
In order to assess the expression pattern of the LPAAT promoter in different tissues, we performed Agrobacterium-mediated transformation with the constructed vectors, which contained different 5′-deletion fragments of LPAAT promoters fused into a gus gene. In order to determine whether the promoter directs the endosperm-specific expression of the target gene, the promoter fragments extending to −896, −817 and −453 were fused to the gus reporter gene, respectively (Fig. 2). These deletions were designed based on the locations of putative elements in the LPAAT upstream region. The constructions were confirmed by PCR and subsequent sequence analysis.

a Schematic representation of the expression vector of LPAAT gene promoter and gus gene constructs. b Details of the translational and transcriptional fusions
Generation of transgenic plants and DNA assay
After the transformation, 37, 42, 39 and 33 transformants were obtained from the constructions pBI101.3-L3, pBI101.3-L2, pBI101.3-L1 and pBI101.3-L0, respectively. Subsequently, we identified 30, 32, 33 and 25 transgene-positive transformants by PCR analysis, respectively. Figure 3 shows that the copy number of the transgenic plants varied from 1 to 4 in the Southern blot analysis. Our data indicated that various LPAAT promoter fragments/GUS fusions were stable inserts in the rice genome.

Southern blot analysis of total genomic DNA of T0 transgenic plants. The DNA samples were digested with HindIII and hybridized with the prepared radioactive probe. The probe size was 0.7 kb and was a fragment of gus gene. M DNA marker, NT Zhonghua 11, and lanes 1–11 transgenic plants
Histological expression analysis
In 2008, all of the T0 transgenic plant (transgenic-positive transformants) tissues were assayed by histochemical staining, respectively. Our data indicated that LPAAT promoter-directed expression of GUS (initiation codon ATG is provided by GUS) was absent in the floral, stalk, root and leaf sheath tissue (Fig. 4e–i) at different developmental stages (data not shown). In contrast, the three LPAAT promoter fragments could generate gus expression in mature seeds, while no GUS expression was observed in the seeds of plants transformed with a promoterless reference plasmid (pBI101.3, Fig. 4a). In particular, GUS expression was not observed in the embryo, but was observed in the endosperm (Fig. 4b–d). For each construct, most of the positive transformants follow the same expression pattern. The ratios are 26/30, 27/32, 25/33, respectively. We found that the expression levels of gus gene directed by different 5′-deletions were different, suggesting the existence of some functional regulatory elements in these fragments. In addition, the transgenic plants with pBI101.3-L3 showed the highest GUS expression level. These findings indicated that gus gene directed by LPAAT promoter was stably expressed in an endosperm-specific manner. Based on the histochemical assay, for each construct, five families with high expression level of GUS were selected for quantitative analysis.

Histochemical localization of GUS activity driven by the LPAAT-promoter and its deletions in matured seeds of transgenic rice. En endosperm, Em embryo. a Negative control seeds transformed with promoterless pBI101.3. b Transgenic seeds with pBI101.3-L1. c Transgenic seeds with pBI101.3-L2. d Transgenic seeds with pBI101.3-L3. e Rootlet of transgenic rice with pBI101.3-L3. f Leaf sheath of transgenic rice with pBI101.3-L3. g Flower of transgenic rice with pBI101.3-L3. h Leaves of transgenic rice with pBI101.3-L3. i Stem of transgenic rice with pBI101.3-L3
Quantitative developmental analysis
In 2009, the selected T1 transgenic plant families were grown on the experimental farm of the Huazhong Agricultural University in Wuhan, China. We obtained developing endosperm tissues in transgenic plants containing different LPAAT promoter fragments (or a promoterless vector) at 10, 15 and 20 DAF, respectively, and then measured the GUS activity by fluorometric determination. In general, the average GUS expression of LD3 (pBI101.3-L3), LD2 (pBI101.3-L2), and LD1 (pBI101.3-L1) transgenes in endosperm was 8.2-, 4.8- and 3.2-fold higher than that in promoterless controls (Fig. 5). The GUS expression of the three different transgenic lines was different during this developmental period. The highest expression level of GUS was at 15 DAF in LD3 and LD2, while the same level was at 10 DAF in LD1. Removal of approximate 443 bp between pBI101.3-L3 and pBI101.3-L1 resulted in a two third loss of GUS activity, suggesting the existence of positive elements driving the expression in this region. Figure 5 shows that the expression driven by the whole fragment was up to 1.76- and 2.8-fold higher than those driven by the −817 bp and −453 bp upstream fragments, and the expression was 10.7-fold higher than that of the promoterless vector.

The quantitative analysis of GUS activity in developing endosperm tissues at 10, 15, and 20 DAF (days after flowering), respectively. 101.3 Control plasmid, LD1 pBI101.3-L1, LD2 pBI101.3-L2, LD3 pBI101.3-L3
Discussion
In this study, we cloned the LPAAT promoter in coconut and investigated its expression pattern based on stable genetic transformation in transgenic rice plantlets. Using histochemical staining of gus gene expression driven by LPAAT promoter, we found that the stable GUS expression was specially localized in the endosperm of transgenic rice seeds, while no GUS expression was detected in floral, stalk, root and leaf sheath tissue. To our knowledge, this is the first promoter sequence from a palmaceous plant species that was characterized and used for cereal genetic modification. A previous study shows that a promoter from a monocotyledonous species has different expression patterns when it is tested in other monocotyledonous species (Furtado et al. 2008). In contrast, the effective expression of gus gene in our study indicates that LPAAT promoter has acceptable universal properties.
The endosperm-specific expression has been investigated in a number of cereal plants, but most of these studies have mainly focused on the expression of storage proteins such as zein (Giovinazzo et al. 1992), glutenin (Thomas and Flavell 1990), glutelin (Kawakatsu et al. 2008) and hordein (Muller and Knudsen 1993). Although the aim in most studies is to increase the carbohydrate and nitrogen content of endosperm (Joersbo et al. 2001), it is also desirable to identify endosperm-specific promoters that respond to the process of fatty acid metabolism (Rasmussen and Donaldson 2006). Most of the known endosperm-specific promoters from monocots are active in the late phases of grain development, and they are mainly expressed in starchy endosperm (Qu and Takaiwa 2004). However, LPAAT is a pivotal enzyme controlling the metabolic flow of lysophosphatidic acids into different phosphatidic acids in various tissues. In higher plants, the subcellular compartments of glycerolipid synthesis are mainly in the endoplasmic reticulum (ER). Glycerol-3-phosphate (GP) is sequentially acylated with acyl-CoA to LPA and PA, which is also catalyzed by glycerol-3-phosphate acyltransferase (GPAT) and LPAAT, respectively. Therefore, the LPAAT promoter is the first tissue-specific promoter responding to fatty acid metabolism in endosperm cells. The special function and subcellular compartment of this LPAAT promoter make it a unique promoter candidate in crop endosperm modification. This promoter offers the potential to improve the rice quality by modifying the quality and quantity of nutrient uptake though the endosperm.
Instead of transient expression analysis, we applied Agrobacterium-mediated stable transformation to validate the function of LPAAT promoter. While transient transformation in specific plant tissues by ballistic transformation is a useful approach to assess the activity of various endosperm-specific promoters (Ahlandsberg et al. 2005; Hwang et al. 2004), Kluth et al. (2002) showed that the transient expression of some tissue-specific promoters can not completely reflect the expression pattern in stable transformants. Furthermore, transient expression studies in endosperm tissues may help elucidate the required promoter sequences, but few studies have shown tissue specificity, relative promoter strength or developmental program as observed for the endogenous genes in the endosperm (Muller and Knudsen 1993). Frisch et al. (1995) showed that chromosomal integration is critical for proper regulation of seed-active promoter by comparing the transient and stable expression of the seed promoter. Therefore, it is more reliable to examine the stable expression of gus gene directed by LPAAT promoter after genetic transformation and plant regeneration in rice.
In the present study, we detected different GUS expression patterns controlled by three deletion fragments without quantification. While GCN4, AACA and G-box or endosperm motif (E-motif) are known to be crucial for endosperm-specific expression in rice (Furtado et al. 2008) and transgenic tobacco (Washida et al. 1999), latent key E-motif in the LPAAT promoter is still unknown. Further studies should focus on the spatial and temporal control of LPAAT promoter and other homologous endosperm-specific promoters in rice. Moreover, in order to completely characterize LPAAT promoter, it is essential to determine the cellular and subcellular localizations of GUS expression under this promoter in rice seeds as well as to compare with the expression level under other endosperm-specific promoters.
Abbreviations
- CTAB:
-
Cetyltriethylammnonium Bromide
- GUS:
-
β-glucuronidase
- LPA:
-
Lysophosphatidic acid
- LPAAT:
-
(1-acyl-sn-glycerol-3-acyltransferase)
- MU:
-
4-Methylumbelliferone
- MUG:
-
4-Methylumbelliferyl-β-d-glucuronide
- PA:
-
Phosphatidic acid
- TGA:
-
Triacylglycerol
- X-Gluc:
-
5-bromo-4-chloro-3-indolyl-β-d-glucuronide
References
Ahlandsberg S, Sun C, Jansson C (2005) An intronic element directs endosperm-specific expression of the sbellb gene during barley seed development. Plant Cell Rep 20:864–868
Bajaj S, Mohanty A (2005) Recent advances in rice biotechnology—towards genetically superior transgenic rice. Plant Biotechnol J 3:275–307
Bhullar S, Chakravarthy S, Advani S, Datta S, Pental D, Burma PK (2003) Strategies for development of functionally equivalent promoters with minimum sequence homology for transgene expression in plants: cis-elements in a novel DNA context versus domain swapping. Plant Physiol 132:988–998
Bradford M (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein–dye binding. Anal Biochem 72:248–254
Charrier B, Scollan C, Ross S, Zubko E, Meyer P (2000) Co-silencing of homologous transgenes in tobacco. Mol Breed 6:407–419
Choi HW, Lemaux PG, Cho MJ (2003) Long-term stability of transgene expression driven by barley endosperm-specific hordein promoters in transgenic barley. Plant Cell Rep 21:1108–1120
Coleman RA, Lee DP (2004) Enzymes of triacylglycerol synthesis and their regulation. Prog Lipid Res 43:134–176
Daniel ZV, Miguel FB, Nelson TH, Patricia CG (2005) Morphological variation of fruit in Mexican populations of Cocos nucifera L. (Arecaceae) under in situ and ex situ conditions. Genet Resour Crop Evol 52:421–434
Frisch DA, van der Geest AHM, Dias K, Hall TC (1995) Chromosomal integration is required for spatial regulation of expression from the β-phaseolin promoter. Plant J 7:503–512
Furtado A, Henry RJ, Takaiwa F (2008) Comparison of promoters in transgenic rice. Plant Biotechnol J 6:679–693
Giovinazzo G, Manzocchi LA, Bianchi MW, Coraggio I, Viotti A (1992) Functional analysis of the regulatory region of a zein gene in transiently transformed protoplasts. Plant Mol Biol 19:257–263
Hiei Y, Ohta S, Komari T, Kumashiro T (1994) Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of boundaries of the T-DNA. Plant J 6:271–282
Hiroi T, Takaiwa F (2006) Peptide immunotherapy for allergic diseases using a rice-based edible vaccine. Curr Opin Allergy Clin Immunol 6:455–460
Huang JQ, Wei ZM, An HL, Zhu YX (2001) Agrobacterium tumefaciens-mediated transformation of rice with the spider insecticidal gene conferring resistance to leaf folder and striped stem borer. Cell Res 11:149–155
Hwang YS, Ciceri P, Parsons RL, Moose SP, Schmidt RJ, Huang N (2004) The maize O2 and PBF proteins act additively to promote transcription from storage protein gene promoters in rice endosperm cells. Plant Cell Physiol 45:1509–1518
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3907
Joersbo M, Marcussen J, Brunstedt J (2001) In vivo modification of the cell wall polysaccharide galactomannan of guar transformed with a α-galactosidase gene cloned from senna. Mol Breed 7:211–219
Kawakatsu T, Yamamoto MP, Hirose S, Yano M, Takaiwa F (2008) Characterization of a new rice glutelin gene GluD-1 expressed in the starchy endosperm. J Exp Bot 59:4233–4245
Kim HU, Huang AH (2004) Plastid lysophosphatidyl acyltransferase is essential for embryo development in Arabidopsis. Plant Physiol 134:1206–1216
Kluth A, Sprunck S, Becker D, Lorz H, Lutticke S (2002) 5′ deletion of a gbss1 promoter region from wheat leads to changes in tissue and developmental specificities. Plant Mol Biol 49:669–682
Knutzon DS, Lardizabal KD, Nelsen JS, Bleibaum JL, Davies HM, Metz JC (1995) Cloning of a coconut endosperm cDNA encoding a 1-acyl-sn-alycerol-3-phosphate acyltransferase that accepts medium-chain-length substrates. Plant Physiol 109:999–1006
Lamacchia C, Shewry PR, Fonzo ND, Forsyth JL, Harris N, Lazzeri PA, Napier JA, Halford NG, Barcelo P (2001) Endosperm-specific activity of a storage protein gene promoter in transgenic wheat seed. J Exp Bot 52:243–250
Li DD, Fan YM (2008) Construction and characterization of a cDNA library from the pulp of coconut (Cocos nucifera L.). Agric Sci China 7:101–105
Li M, Singh R, Bazanova N, Milligan AS, Shirley N, Langridge P, Lopato S (2008) Spatial and temporal expression of endosperm transfer cell-specific promoters in transgenic rice and barley. Plant Biotechnol J 6:465–476
Lin YJ, Zhang Q (2005) Optimizing the tissue culture conditions for high efficiency transformation of indica rice. Plant Cell Rep 23:540–547
Liu XL, Tang H, Li DD, Wang Q, Lin YQ, Zhou R (2008) Genetic diversity of coconut cultivars from Hainan province on SSR markers. Acta Hortic Sin 35:1199–1204
Muller M, Knudsen S (1993) The nitrogen response of a barley C-hordein promoter is controlled by positive and negative regulation of the GCN4 and endosperm box. Plant J 4:343–355
Nath UK, Wilmer JA, Wallington EJ, Becker HC, Möllers C (2009) Increasing erucic acid content through combination of endogenous low polyunsaturated fatty acids alleles with Ld-LPAAT + Bn-fae1 transgenes in rapeseed (Brassica napus L.). Theor Appl Genet 118:765–773
Okazaki K, Sato N, Tsuji N, Tsuzuki M, Nishida I (2006) The significance of C16 fatty acids in the sn-2 positions of glycerolipids in the photosynthetic growth of Synechocystis sp. PCC6803. Plant Physiol 141:546–556
Qu LQ, Takaiwa F (2004) Evaluation of tissue specificity and expression strength of rice seed component gene promoters in transgenic rice. Plant Biotechnol J 2:113–125
Qu LQ, Xing YP, Liu WX, Xu XP, Song YR (2008) Expression pattern and activity of six glutelin gene promoters in transgenic rice. J Exp Bot 59:2417–2424
Rasmussen TB, Donaldson IA (2006) Investigation of the endosperm-specific sucrose synthase promoter from rice using transient expression of reporter genes in guar seed tissue. Plant Cell Rep 25:1035–1042
Rivera R, Edwards KJ, Barker JHA, Arnold GM, Ayad G, Hodgkin T (1999) Isolation and characterization of polymorphic microsatellites in Cocos nucifera L. Genome 42:668–675
Russell DA, Fromm ME (1997) Tissue-specific expression in transgenic maize of four endosperm promoters from maize and rice. Transgenic Res 6:157–168
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual, 2nd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Takaiwa F, Takagi H, Hirose S, Wakasa Y (2007) Endosperm tissue is a good production platform for artificial recombinant proteins in transgenic rice. Plant Biotechnol J 5:84–92
Thomas MS, Flavell RB (1990) Identification of an enhancer element for the endosperm-specific expression of high molecular weight glutenin. Plant Cell 2:1171–1180
Washida H, Wu CY, Suzuki A, Yamanouchi U, Akihama T, Harada K, Takaiwa F (1999) Identification of cis-regulatory elements required for endosperm expression of the rice storage protein glutelin gene Glub-1. Plant Mol Biol 40:1–12
Wiley PR, Tosi P, Evrard A, Lovegrove A, Jones HD, Shewry PR (2007) Promoter analysis and immunolocalisation show that puroindoline genes are exclusively expressed in starchy endosperm cells of wheat grain. Plant Mol Biol 64:125–136
Wu J, Yu L, Li L, Hu J, Zhou J, Zhou X (2007) Oral immunization with transgenic rice seeds expressing VP2 protein of infectious bursal disease virus induces protective immune responses in chickens. Plant Biotechnol J 5:570–578
Acknowledgments
This research was supported by the National Natural Science Foundation of China (NSFC) (No: 30560092 and 30860149), the National High Technology Research and Development Program of China (863 Program) and National Nonprofit Institute Research Grant of CATAS-ITBB.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Communicated by M. Jordan.
L. Xu and R. Ye contributed equally to the work.
Rights and permissions
About this article
Cite this article
Xu, L., Ye, R., Zheng, Y. et al. Isolation of the endosperm-specific LPAAT gene promoter from coconut (Cocos nucifera L.) and its functional analysis in transgenic rice plants. Plant Cell Rep 29, 1061–1068 (2010). https://doi.org/10.1007/s00299-010-0892-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-010-0892-y