
Abstract
Direct genetic transformation of mesophyll protoplasts was studied in Pelargonium × hortorum. Calcein and green-fluorescent protein (GFP) gene were used to set up the process. Electroporation (three electric pulses from a 33-μF capacitor in a 250-V cm−1 electric field) was more efficient than PEG 6000 for membrane permeation, protoplast survival and cell division. Transient expression of GFP was detected in 33–36% of electroporated protoplasts after 2 days and further in colonies. A protoplast suspension conductivity of >1,500 μS cm−1 allowed high colony formation and plant regeneration. Stable transformation was obtained using the plasmid FAJ3000 containing uidA and nptII genes. When selection (50 mg l−1 kanamycin) was achieved 6 weeks after electroporation, regenerated shoots were able to grow and root on 100 mg l−1 kanamycin. The maximum transformation efficiency was 4.5%, based on the number of colonies producing kanamycin-resistant rooted plants or 0.7% based on the number of cultured protoplasts. Polymerase chain reaction (PCR) analysis on in vitro micropropagated plants showed that 18 clones out of 20 contained the nptII gene, while the uidA gene was absent. These results were confirmed after PCR analyses of five glasshouse-acclimatized clones.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Pelargonium × hortorum L. H. Bailey is the most economically important bedding and pot plant in Europe and North America (Mithila et al. 2001). Development of efficient genetic transformation systems can provide opportunities to improve P. × hortorum for characteristics such as novel flower colours or resistance to biotic and abiotic stresses. For these purposes, we previously set up an efficient transformation system using leaf discs and Agrobacterium tumefaciens (Hassanein et al. 2005). Transformation of Pelargonium using direct gene transfer is another possibility, since protoplast technology is now well known for P. × hortorum (Nassour and Dorion 2002; Nassour et al. 2003; Hassanein and Dorion 2006). In this process, the micro-callus formation frequency reached 31–40% and regeneration frequency was up to 65–83% of the calli (Hassanein and Dorion 2006). Naked DNA can be introduced into protoplasts in which membrane permeability was increased either by chemicals such as polyethylene glycol (PEG) or by electric pulses (electroporation) (Davey et al. 2005). Electroporation is the most commonly used technique because it is known to be rapid, simple, efficient and non-toxic. Transformation efficiency depends on factors such as protoplast density and viability, cell susceptibility to electroporation, medium osmolarity, electroporation buffer, DNA concentration, electric field voltage and capacitance (Prasanna and Panda 1997; Fisk and Dandekar 2004). For PEG-mediated direct transformation, the amount of plasmid, PEG concentration and inoculation time are the most important factors affecting DNA uptake in many plant species (Rasmussen and Rasmussen 1993; Locatelli et al. 2003). We report here, on the regeneration of transgenic P. × hortorum via direct gene transfer into protoplasts. Comparisons were made between the two direct transformation methods (PEG and electroporation). Optimum conditions for transient gene expression were determined by measuring calcein uptake in the protoplasts as reported for grapevine and tobacco (Montane et al. 1990; Valat et al. 2000). Green-fluorescent protein (GFP) gene was also used since transient expression and stable transformation were reported in many plant species (Hu and Cheng 1995; Winfield et al. 2001). Finally, stable transformation was studied using the plasmid already used in the laboratory (Hassanein et al. 2005), precisely pFAJ3000 containing uidA and nptII genes (De Bondt et al. 1994).
Materials and methods
Isolation and culture of mesophyll protoplasts
Protoplasts were isolated from in vitro grown plants of P. × hortorum ‘Panaché Sud’ according to Hassanein and Dorion (2006). After two washings by centrifugation (Nassour and Dorion 2002) in CPW solution (Frearson and Power 1973) containing 0.5 M mannitol, protoplasts were rinsed one to two times in the electroporation buffer (MKM) containing 0.5 M mannitol, 5 mM KCl and 200 μM MOPS (pH 7.2) to eliminate salt traces (Guerche et al. 1987a, b). Protoplasts were finally suspended in this buffer at a density of 2 × 106 protoplasts ml−1 for electroporation, or in culture medium at 4 × 106 protoplasts ml−1 for PEG treatment. Protoplasts were plated at a final density of 3 × 104 protoplasts ml−1 in the liquid culture medium. All the steps of culture process were performed according to Hassanein and Dorion (2006). After 1.5–2 months, microcalli reached 0.5–1-mm diameter and were subcultured individually. The callus needed 3–4 more weeks to reach a size of 5–7-mm diameter. Shoot regeneration, elongation, rooting and glasshouse acclimatization were performed as described previously (Hassanein and Dorion 2006).
Determination of permeation conditions using calcein
A solution of 75 mM calcein (C30H26N2O13) was prepared, then filter sterilized (0.22 μm) before using in transfection experiments.
For PEG-mediated calcein uptake, 35 μl of calcein was mixed, in a test tube, with 200 μl of protoplast suspension (4 × 106 protoplasts ml−1). After 5 min, a sterile solution containing 40% PEG 6000, 0.8 M sorbitol or 0.5 M mannitol, 100 mM Ca (NO3)2 and 17.5 mM MES (pH 6) was added. PEG concentration in this solution (25 or 40% taking in account its osmolarity), solution volume added to protoplast suspension (150 or 200 μl) and length of treatment (10 or 20 min) were studied. After PEG treatment, protoplasts were rinsed twice in CPW solution containing 0.5 M mannitol and centrifuged for 10 min at 60 × g to eliminate PEG and calcein before counting. Finally, protoplasts were washed and collected in culture medium.
Electroporation of protoplasts was adapted from Guerche et al. (1987a) and Chupeau et al. (1994). The protoplast suspension in MKM (2 × 106 protoplasts ml−1), calcein and 1 M KCl sterile solution were incubated on ice for 1 h before electroporation. The electroporation chamber was first sterilized with 70% ethanol and washed with sterile MKM solution before addition of 1 ml of protoplast suspension. After addition of calcein, the conductivity of the protoplast suspension was adjusted to 730 or 1,850 μS cm−1, using KCl solution. The electric field (250 or 300 V cm−1) and different capacities (10, 33 or 50 μF) were also studied. Three electric pulses of 10 s, to be sure that the capacity was totally discharged, were delivered at 20-s interval. Treated protoplasts were rinsed and collected in culture medium as described previously for PEG treatment.
For both methods, 3 ml of protoplast suspension were cultured per Petri dish according to Hassanein and Dorion (2006). The plating density was adjusted to 5 × 104 protoplasts ml−1, taking into account that it could decrease to 3 × 104 protoplasts ml−1 after mortality caused by treatments.
Plasmids
Two plasmids were used in electroporation experiments. The pEGFP (BD Biosciences, Clontech) harbours the enhanced GFP (EGFP) driven by the cauliflower mosaic virus (CaMV) 35S promoter and terminator. The pFAJ3000 carries the neomycin phosphotransferase II (nptII) gene as selectable marker under the control of the nopaline synthase promoter and octopine synthase terminator, and the β-glucuronidase (uidA) gene with an intron under the control of 35S promoter and terminator (De Bondt et al. 1994). Plasmid DNA was purified from overnight cultures of Escherichia coli using NucleoBond AX 500 kit for purification of plasmid DNA (Macherey-Nagel, France) according to the manufacturer’s instructions. The plasmids were maintained at −20°C, precipitated in ethanol, until use. For electroporation, the plasmids were suspended in MKM electroporation buffer.
Electroporation using pEGFP
Two conditions of electroporation were evaluated using pEGFP. As much as 15–20 μg of plasmid DNA was added to the electroporation chamber containing 1 ml of protoplasts (2 × 106 protoplasts ml−1) previously maintained on ice for 1 h. The conductivity was left without change (850 μS cm−1) or adjusted to (1,540 μS cm−1) using KCl. Three electric pulses were delivered as previously mentioned, by a 33-μF capacitor at 250 V cm−1.
Electroporation using pFAJ3000
Before electroporation using pFAJ3000, the sensitivity of protoplasts to paromomycin sulfate (Sigma) and kanamycin monosulphate (Sigma) was studied. Two concentrations of paromomycin (10 or 20 mg l−1) and four of kanamycin (10, 20, 50 or 100 mg l−1) were studied at four different stages: division (2–8 cells) after 10 days of culture, microcolony (32–64 cells) after 4 weeks of culture, callus after 6–8 weeks of culture, and shoot regeneration stage. In both the first stages, the selective agent was added at the final desirable level when the medium was renewed (Hassanein and Dorion 2006). For the rooting stage, the susceptibility to 100 mg l−1 kanamycin was already known (Hassanein et al. 2005).
As much as 20–25 μg of pFAJ3000 was mixed with 1 ml protoplast suspension (2 × 106 protoplast ml−1). The conductivity was adjusted to 1,790 μS cm−1. Electric pulses (33 μF at 250 V cm−1) were delivered as described for pEGFP. Transformed colonies were selected first at division or microcolony stages using paromomycin (20 mg l−1) or kanamycin (50 or 100 mg l−1). Two weeks after selection, all colonies were spread on gelled medium (2 g l−1 gelrite, Merck) without selection for 1 week. Colonies were then transferred to callus medium under selection pressure. In some cases, the selection was not applied before the callus stage.
All selected calli were subcultured on the regeneration medium containing 100 mg l−1 kanamycin for 4 weeks and finally transferred to elongation medium containing 100 mg l−1 kanamycin for another 4 weeks. One single shoot was isolated from each callus and transferred to selective rooting medium (100 mg l−1 kanamycin). Plants were maintained under selection pressure for approximately 6 months before acclimatization in a glasshouse designated for GMOs.
Control of genetic transformation
GUS histochemical assay
β-Glucuronidase expression was assayed on leaves of kanamycin-resistant rooted plants in vitro. Leaf discs and young leaves of rooted plants were stained by the GUS histochemical staining method as described by Jefferson et al. (1987) with some modifications. Samples were incubated overnight at 37°C in a solution containing 3 mM 5-bromo-4-chloro-3-indolyl-B-d-glucuronic acid (X-Gluc), 4 mM potassium ferricyanide, 0.05 mM potassium ferrocyanide, 10 mM EDTA and 50 mM of sodium phosphate buffer (pH 7.2). Chlorophyll was cleared by soaking samples in 70% (v/v) ethanol with agitation for 3–4 h.
DNA isolation and polymerase chain reaction analysis
Genomic DNA was isolated from young fully expanded leaves of control and kanamycin-resistant rooted plants using the CTAB method (Briard et al. 2000) for in vitro plants, or the Nucleon extraction Kit (GE Healthcare, Amersham Biosciences) according to the manufacturer’s protocol for 1-year acclimatized plants. The sequence of primers used for GUS (uidA) and kanamycin (nptII) amplification have been previously described (Hassanein et al. 2005):
-
uidA forward primer, 5′-GGTGGGAAAGCGCGTTACAAG-3′,
-
uidA reverse primer, 5′-GTTTACGCGTTGCTTCCGCCA-3′,
-
nptII forward primer, 5′-GAGGCTATTCGGCTATGACTG-3′,
-
nptII reverse primer 5′-ATCGGGAGCGGCGATACCGTA-3′.
The expected fragment lengths were 1,200 and 700 bp for uidA and nptII, respectively. The polymerase chain reaction (PCR) amplification reactions of 10 μl contained 40–90 ng DNA, 0.15–0.22 μM of each primer, 1.5 mM MgCl2, 200 μM dNTP, 1 × Taq polymerase reaction buffer and 0.25 U Taq polymerase (Silverstar or UptiTherm). Thermocycling was carried out according to Hassanein et al. (2005). Amplified fragments were analysed by electrophoresis at 120 V for 60 min on 1.4% agarose gel (DNA of in vitro grown plants) or 100 V for 30 min on 1.6% agarose gel (DNA of greenhouse grown plants), followed by colouration with ethidium bromide and detection under UV illumination.
Data analysis
The efficiency of PEG and electroporation treatments were evaluated via calcein uptake measurement as the percentage of viable green fluorescing protoplasts (Olympus microscope BH2) and protoplast survival (%) measured immediately, or 48 h after transfection. Cell division and microcolony formation (%) were evaluated after 1–4 weeks in culture medium. The transient expression of GFP was detected under blue or UV light (450–490 nm) and the efficiency was evaluated as above. Percentage of shoot regeneration was also determined (percentage of calli inducing buds or shoots after 4 weeks on regeneration medium). Transformation efficiency, using pFAJ3000, was calculated as the ratio of resistant colonies to total number of cultured protoplasts, and as the ratio of kanamycin-resistant rooted plants to the number of tested colonies × 100.
In experiments using pEGFP and pFAJ3000, two controls were added, protoplasts without electroporation (control-1) and electroporated protoplasts without plasmid (control-2). Each experiment (four Petri dishes per treatment) was repeated twice. For survival, calcein uptake, division and colony formation, counts were carried out on 150–300 cells per Petri dish. Proportions were compared using G test of independence based on Chi-square tables (Sokal and Rohlf 1981) and significance was tested at P ≤ 0.05. Otherwise, means were given with confidence interval at P = 0.05.
Results
Determination of optimal permeation conditions using calcein
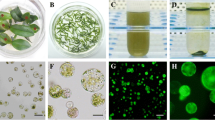
After PEG treatments, protoplast survival dropped significantly whatever the conditions (Table 1). Calcein uptake reached 25.6–38.1% when 200 μl of 40% PEG solution was added to the protoplast suspension, whether the treatment lasted 10 or 20 min, but no division occurred after 10 days (Table 1, Fig. 1a). Divisions were only observed (≈40% of the control) when 150 μl of 25% PEG solution was used, but calcein uptake was significantly lower (Table 1). Electroporation was better than PEG treatment (Table 2), since greater survival (74–83%), division percentage and microcolony formation were obtained, whatever the treatment. The highest percentage of calcein uptake was obtained using an electric field of 250 V cm−1 and a capacitance of 33 μF regardless of the initial conductivity of protoplast suspension (Fig. 1b). After 48 h, the protoplast viability was approximately 50–60 versus 83% for control-1 (without electroporation). Further, detection of calcein in dividing cells and microcolonies was difficult due to dilution during division and chlorophyll masking. Nevertheless, the best conditions were retained for further experiments.

Calcein uptake in Pelargonium × hortorum ‘Panaché Sud’ mesophyll protoplasts following PEG or electroporation treatment. a Calcein uptake after PEG treatment, b calcein uptake after electroporation (bar 30 μm)
Determination of optimal conductivity using GFP expression
Electroporation was performed on protoplast suspensions at low (850 μS cm−1) and high (1,540 μS cm−1) conductivity (Table 3). In the first case, conductivity increased during electric pulses from 850 to 980 μS cm−1, whereas it remained stable in the second case (1,540–1,560 μS cm−1). Viability after treatment decreased significantly when conductivity was low (Table 3). Green fluorescence was observed in both conditions (Table 3, Fig. 2a), but only in viable protoplasts (Fig. 2a), while the control showed only the red fluorescence of chlorophyll. The percentage of division was greater significantly for electroporated protoplasts than for control (Table 3). However, fewer and smaller colonies resulted from electroporated protoplasts at low conductivity. GFP expression was detected either in dividing protoplasts, 10 days after electroporation, or in colonies after 4 weeks (Fig. 2b, c). Quantification of GFP stable expression was impossible due to masking by chlorophyll fluorescence. Calli and shoots were obtained from control and both electroporation treatments (Fig. 2d); but high conductivity treatment was still significantly better than low conductivity treatment (Table 3). It was retained for further experiments.

Electroporation-mediated GFP uptake into Pelargonium × hortorum ‘Panaché Sud’ mesophyll protoplasts. a Green fluorescence of GFP in electroporated protoplasts and red fluorescence of chlorophyll (bar 30 μm); b divided protoplasts showing GFP gene expression after 10 days of electroporation (bar 60 μm); c colony expressing GFP gene after 4 weeks of culture (bar 30 μm). d Shoot regeneration from electroporated protoplast-derived calli (bar 1 cm)
Stable transformation by electroporation of protoplasts with pFAJ 3000
Cell susceptibility to neomycins
After 10 days of culture, 46.4% of viable protoplasts divided. The addition of 20 mg l−1 paromomycin or 50–100 mg l−1 kanamycin, at this stage, resulted in necrosis of all dividing and non-dividing cells within 2 weeks. All the other conditions allowed some divisions without microcolony development. Similar results were obtained when antibiotics were applied after 4 weeks of culture (Table 4). However, some microcolonies were observed at the lowest doses such as 10 mg l−1 paromomycin and 10 or 20 mg l−1 kanamycin. In contrast, when antibiotics were added at the callus stage, callogenesis occurred in all conditions, except 50 and 100 mg l−1 kanamycin (Table 4). When antibiotics were applied in the regeneration medium, 100 mg l−1 kanamycin induced callus necrosis (Table 4). All other conditions allowed shoot growth, but regeneration was significantly less than that of the control (Table 4).
Selection after electroporation
When electroporation was performed using pFAJ3000, the viability of protoplasts was high and significantly more divisions were observed, compared to control (Table 5, Fig. 3a). Three weeks after treatment, protoplasts formed microcolonies. Application of the selection pressure, 1 week later with 50 mg l−1 kanamycin allowed the recovery of resistant colonies after 2 weeks (Table 5, Fig. 3b). A total of 51 colonies were recovered per Petri dish and formed calli. No control colonies survived under similar selection pressure. When selection pressure was applied 6–8 weeks after electroporation (Table 6), calli developed on all selection media. Kanamycin seemed more selective (Table 6) since calli formed on paromomycin were green, while those growing on kanamycin were white in colour (Figure 3c, d).

Stable transformation of P. × hortorum ‘Panaché Sud’ after electroporation. a Divided protoplasts 1 week after electroporation (bar 30 μm). b Colonies selected with 50 mg l−1 kanamycin added 4 weeks after electroporation (bar 5 mm). c Electroporated protoplasts-derived green calli on selection medium containing 20 mg l−1 paromomycin (bar 5 mm). d Calli obtained on selection medium containing 50 mg l−1 kanamycin (bar 5 mm). e Shoot regeneration on selection medium containing 100 mg l−1 kanamycin (bar 5 mm). f Plant rooted on selection medium containing 100 mg l−1 kanamycin (bar 10 mm)
When resistant calli were transferred to regeneration medium containing 100 mg l−1 kanamycin, some green shoots were obtained (Fig. 3e) mainly when initial selection was performed with 10 and 20 mg l−1 kanamycin or paromomycin (Table 6). On such selection pressure, 22 kanamycin-resistant rooted plants were obtained (Fig. 3f). Regeneration was not obtained from electroporated control (control-2) calli on similar selection medium. Transformation efficiency based on kanamycin-resistant rooted plants varied between 0.7 and 4.5% and was higher when initial selection was performed with 10 and 20 mg l−1 kanamycin or paromomycin (Table 6). A total of 20 clones were finally submitted to DNA analysis. All of them were preserved at the in vitro stage and 5 out of 20 were acclimatized in the greenhouse for DNA extraction 1 year later.
GUS expression in kanamycin-resistant plants
GUS expression was histochemically examined in kanamycin-resistant rooted plants grown in vitro. GUS activity was never detected in control plants or in the kanamycin-resistant plants.
PCR analysis of kanamycin-resistant plants
The DNA of kanamycin-resistant plants of P. × hortorum (grown in vitro and in glasshouse) was subjected to PCR analyses. For in vitro-grown plants, 18 plants out of 20 showed the expected band for the nptII gene (data not shown). One year later, all the five clones grown in the glasshouse and already tested before acclimatization also showed the expected band for nptII gene (Fig. 4). None of the tested plants showed the 1,200 bp band for uidA gene.

Polymerase chain reaction analysis for detecting nptII gene in plants regenerated after electroporation in vitro of leaf protoplasts Pelargonium × hortorum ‘Panaché Sud’ with pFAJ 3000. DNA was extracted from greenhouse-grown plants 8 months after acclimatization. L SmartLadder (EUROGENTEC, 200 lanes MW-1700-02), P pFAJ3000 plasmid (white arrow indicates nptII gene 700 bp), C control clone arising from non-electroporated protoplast. 4, 5, 8, 11, 20 Pelargonium × hortorum ‘Panaché Sud’ putative transgenic clones
Discussion
In this study, we determined the optimum conditions for direct genetic transformation of leaf protoplasts of P. × hortorum ‘Panaché Sud’. Pelargonium plants resistant to kanamycin were obtained for the first time through protoplast electroporation. PCR analysis confirmed the presence of the nptII gene in these plants. Although we did not perform Southern analysis, it is likely that the main part of these plant are true transformants since (1) pollution by A. tumefaciens is impossible because only plasmid was used, (2) plants were maintained in aseptic conditions for at least five subcultures before acclimatization and (3) in vitro or glasshouse control plants were negative.
Calcein was used to study membrane permeation via PEG or electroporation treatment. This non-permeant dye was used previously for setting up the optimal conditions of DNA uptake into grapevine and tobacco protoplasts (Montane et al. 1990; Valat et al. 2000). In our experiments, we found that electroporation was more efficient than PEG treatment, in agreement with results previously reported in rice, wheat and barley protoplasts (Yang et al. 1988; Diaz 1994). Despite the high calcein uptake induced by PEG treatment, survival was low and cell division was prevented. This can be explained by a toxic effect of PEG enhanced by toxic substances released by dying protoplasts (Bhojwani and Razdan 1996).
The parameters determined for protoplast electroporation were 250 V cm−1 and 33 μF. Generally, a capacitor of 15 μF charged at 250 V cm−1 was effective for transformation of many species as reported by Chupeau et al. (1994). The need for greater capacitance for more efficient permeation of P. × hortorum protoplasts may be related to the high conductivity of the protoplast solution.
The positive effect of electroporation on plant regeneration was mentioned in some studies (Chand et al. 1988) and negative effect was found in others (Quecini et al. 2002). In our experiments, protoplast division was stimulated as mentioned by Rech et al. (1987), but regeneration remained unchanged.
Results obtained using pEGFP in electroporation experiments showed the feasibility of transient expression and stable transformation, since fluorescence was still noticeable after 4 weeks. However, transformation efficiency was not determined because of GFP masking by chlorophyll fluorescence (Winfield et al. 2001). Therefore, transformation attempts were made using pFAJ3000 harbouring the neomycin resistance gene. Control protoplasts were highly susceptible to neomycins at early stages of development (10 and 28 days). However, these earliest stages were not usable for selection of transformed colonies as previously shown with lettuce protoplasts (Chupeau et al. 1989). Selection at the 4 weeks stage resulted in limited number of colonies selected against 50 mg l−1 kanamycin (51 out of 15 × 104 cultivated protoplasts per Petri dish), which gave a transformation efficiency of 3.4 × 10−4. This low efficiency might result from the release of toxic substances by kanamycin-susceptible cells, which, in turn, can prevent the development of kanamycin-resistant colonies (Guerche et al. 1987a). Consequently, selection was applied 6–8 weeks after electroporation as it was done in lettuce protoplasts (Chupeau et al. 1989). Paromomycin (20 mg l−1) gave a high per cent of green calli formation, which resulted in greater regeneration and improved transformation efficiency (2.8%). This result is in agreement with those of Guerche et al. (1987a) and Chupeau et al. (1989), who used the same paromomycin dose as a substitute to kanamycin selection of transformed colonies. More higher regeneration and rooting on kanamycin 100 mg l−1 and consequently higher transformation efficiency up to 3.8–4.5% were obtained when selection pressure was applied with kanamycin levels lower than 50 mg l−1. Transformation efficiency can reach 0.7% based on the number of electroporated protoplasts producing kanamycin-resistant rooted plants. However, stable transformation, in our experiments, was restricted to the gene under selection since all the kanamycin-resistant plants, although transgenic for nptII, were not transgenic for uidA gene. This can be explained by deletions in the plasmid before or after its integration into the plant genome as mentioned in other plant species transformed via A. tumefaciens (Kishimoto et al. 2002; Cui et al. 2004). In a previous study on the same Pelargonium cultivar (Hassanein et al. 2005), 6 plants out of 36 displayed this phenomenon. Therefore, it is likely that the method itself could induce this high level of genetic reorganization, as it was shown by Chupeau et al. (1994) on transgenic poplar that only the gene under selection was integrated in the plant genome. Thus, direct transformation of Pelargonium via electroporation of protoplasts should be useful to transfer and study agronomic traits selectable in vitro, as those for drought or disease resistance.
Abbreviations
- BAP:
-
6-Benzylaminopurine
- CPW:
-
Cell and protoplast washing solution (Frearson and Power 1973)
- GFP:
-
Green-fluorescent protein
- IAA:
-
Indole-3-acetic acid
- MES:
-
2-(N-morpholino)-ethanesulfonic acid
- MKM:
-
Electroporation buffer (Guerche et al. 1987a)
- NAA:
-
α-Naphthaleneacetic acid
- PVP-10:
-
Polyvinyl pyrrolidone (MW, 10,000)
- TDZ:
-
Thidiazuron
References
Bhojwani SS, Razdan MK (1996) Plant tissue culture: theory and practice. Elsevier science, Amsterdam, p 767
Briard M, LeClerc V, Grzebelus D, Senalik D, Simon PW (2000) Modified protocols for rapid carrot genomic DNA extraction and AFLP™ analysis using silver stain or radioisotopes. Plant Mol Biol Rep 18:235–241
Chand PK, Ochatt SJ, Rech EL, Power JB, Davey MR (1988) Electroporation stimulates plant regeneration from protoplasts of the woody medicinal species Solanum dulcamara L. J Exp Bot 39:1267–1274
Chupeau MC, Bellini C, Guerche P, Maisonneuve B, Vastra G, Chupeau Y (1989) Transgenic plants of lettuce (Lactuca sativa) obtained through electroporation of protoplasts. Biotechnology 7:503–508
Chupeau MC, Pautot V, Chupeau Y (1994) Recovery of transgenic trees after electroporation of poplar protoplasts. Transgenic Res 3:13–19
Cui ML, Ezura H, Nishimura S, Kamada H, Handa T (2004) A rapid Agrobacterium-mediated transformation of Antirrhinum majus L. by using direct shoot regeneration from hypocotyl explants. Plant Sci 166:873–879
Davey MR, Anthony P, Power JB, Lowe KC (2005) Plant protoplasts: status and biotechnological perspectives. Biotechnol Adv 23:131–171
De Bondt A, Eggermont K, Druart P, De Vil M, Goderis I, Vanderleyden J, Broekaert WF (1994) Agrobacterium-mediated transformation of apple (Malus × domestica Borkh.): an assessment of factors affecting gene transfer efficiency during early transformation steps. Plant Cell Rep 13:587–593
Diaz I (1994) Optimization of conditions for DNA uptake and transient GUS expression in protoplasts for different tissues of wheat and barley. Plant Sci 96:179–187
Fisk HJ, Dandekar AM (2004) Electroporation: introduction and expression of transgenes in plant protoplasts. Methods Mol Biol 286:79–90
Frearson EM, Power JB (1973) The isolation, culture and regeneration of petunia leaf protoplasts. Dev Biol 33:130–137
Guerche P, Bellini C, Le Moullec JM, Caboche M (1987a) Use of a transient expression assay for the optimization of direct gene transfer into tobacco mesophyll protoplasts by electroporation. Biochimie 69:621–628
Guerche P, Charbonnier M, Jouanin L, Tourneur C, Paszkowski J, Pelletier G (1987b) Direct gene transfer by electroporation in Brassica napus. Plant Sci 52:111–116
Hassanein A, Dorion N (2006) High-efficiency colony formation and whole plant regeneration from mesophyll protoplasts of Pelargonium × hortorum ‘Panaché Sud’. J Hortic Sci Biotechnol 81:714–720
Hassanein A, Chevreau E, Dorion N (2005) Highly efficient transformation of zonal (Pelargonium × hortorum) and scented (P. capitatum) geraniums via Agrobacterium tumefaciens using leaf discs. Plant Sci 169:532–541
Hu W, Cheng CL (1995) Expression of Aequorea green fluorescent protein in plant cells. FEBS Lett 369:331–334
Jefferson RA, Kavanagh TA, Bevan MW (1987) GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J 6:3901–3907
Kishimoto S, Aida R, Shibata M (2002) Agrobacterium tumefaciens-mediated transformation of Elatior Begonia (Begonia × hiemalis Fotsch). Plant Sci 162:697–703
Locatelli F, Vannini C, Magnani E, Coraggio I, Bracale M (2003) Efficiency of transient transformation in tobacco protoplasts is independent of plasmid amount. Plant Cell Rep 21:865–871
Mithila J, Murch SJ, KrishnaRaj S, Saxena PK (2001) Recent advances in Pelargonium in vitro regeneration systems. Plant Cell Tissue Organ Cult 67:1–9
Montane MH, Dupille E, Alibert G, Teissie J (1990) Induction of a long-lived fusogenic state in viable plant protoplasts permeabilized by electric fields. Biochim Biophys Acta 1024:203–207
Nassour M, Dorion N (2002) Plant regeneration from protoplasts of micropropagated Pelargonium × hortorum ‘Alain’: effect of some environmental and medium factors on protoplast system efficiency. Plant Sci 163:169–176
Nassour M, Chassériaux G, Dorion N (2003) Optimization of protoplast-to-plant system for Pelargonium × hortorum ‘Alain’ and genetic stability of the regenerated plants. Plant Sci 165:121–128
Prasanna GL, Panda T (1997) Electroporation: basic principles, practical considerations and applications in molecular biology. Bioprocess Eng 16:261–264
Quecini VM, de Oliveira CA, Alves AC, Vieira MLC (2002) Factors influencing electroporation-mediated gene transfer to Stylosanthes guianensis (Aubl.) Sw. protoplasts. Genet Mol Biol 25:73–80
Rasmussen JO, Rasmussen OS (1993) PEG-mediated DNA uptake and transient GUS expression in carrot, rapeseed and soybean protoplasts. Plant Sci 89:199–207
Rech EL, Ochatt SJ, Chand KP, Power JB, Davey MR (1987) Electro-enhancement of division of plant protoplast-derived cells. Protoplasma 141:169–176
Sokal RR, Rohlf FJ (1981) Tests of independence. W. H. Freeman and Company, San Francisco, pp 731–747
Valat L, Toutain S, Courtois N, Gaire F, Decout E, Pinck L, Mauro MC, Burrus M (2000) GFLV replication in electroporated grapevine protoplasts. Plant Sci 155:203–212
Winfield S, Lawton R, Daniell H, Dhir SK (2001) Transformation of sweet potato tissues with green-fluorescent protein gene. In vitro Cell Dev Biol Plant 37:648–653
Yang H, Zhang HM, Davey MR, Milligan BJ, Cocking EC (1988) Production of kanamycin-resistant rice tissues following DNA uptake into protoplasts. Plant Cell Rep 7:421–425
Acknowledgments
We would like to thank M.C. Chupeau and E. Chevreau from INRA (Institut National de la Recherche Agronomique) for helpful assistance and L. Voisine from ARES (Anjou Recherche Semences) for technical support. We are very grateful to INRA, French Ministry of Agriculture and the Egyptian Government for financial support. We would also like to thank Dr Rolf Deblaere (Bayer Bioscience) for permission to use the pFAJ3000.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by L. Jouanin.
Rights and permissions
About this article
Cite this article
Hassanein, A., Hamama, L., Loridon, K. et al. Direct gene transfer study and transgenic plant regeneration after electroporation into mesophyll protoplasts of Pelargonium × hortorum, ‘Panaché Sud’. Plant Cell Rep 28, 1521–1530 (2009). https://doi.org/10.1007/s00299-009-0751-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-009-0751-x