
Abstract
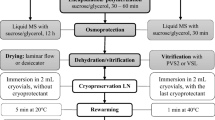
Shoot tips of sweet potato were successfully cryopreserved using an encapsulation vitrification method. Encapsulated shoot tips were pre-incubated in liquid Murashige-Skoog medium containing 30 g/l sucrose for 24 h, then precultured in sucrose-enriched medium (0.3 M sucrose) for 16 h. Shoot tips were osmoprotected with a mixture of 2 M glycerol and 1.6 M sucrose for 3 h before being dehydrated with a highly concentrated vitrification solution (PVS2) for 1 h at 25°C. The encapsulated and dehydrated shoot tips were transferred to a 2 ml cryotube, suspended in 0.5 ml PVS2, and plunged directly into liquid nitrogen. Rapidly warmed shoot tips developed normal shoots and roots in 21 days without any morphological abnormalities after plating on a recovery medium. High levels (average of about 80%) of shoot formation were obtained for three cultivars of sweet potato. This encapsulation vitrification method appears promising for cryopreservation of sweet potato germplasm.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cryopreservation of shoot tips is a logical choice for the long-term storage of germplasm of vegetatively propagated plants, conferring genetic stability with minimum space and maintenance requirements (Bajaj 1978; Harding and Benson 1994; Sakai 1995). Simple and reliable methods for cryopreservation using vitrification (Langis et al. 1990; Sakai et al. 1990) and encapsulation-dehydration (Fabre and Dereuddre 1990) were reported almost 10 years ago. These new procedures dehydrate a major part of the freezable water from the tissues at non-freezing temperatures and enable them to be cryopreserved by being plunged directly into liquid nitrogen (LN). These protocols simplified the cryogenic process and increased the applicability of cryopreservation to a wide range of plant materials, especially non-cold-hardy tropical plants. The vitrification method, with or without encapsulation, has proved to be more effective in producing a higher percentage of survival and faster recovery growth than other cryogenic procedures (Matsumoto et al. 1995a, 1995b; Hirai and Sakai 1999a, 1999b). Over the past decade, the vitrification method has become the preferred method for the cryopreservation, with more than 160 species and cultivars being successfully cryopreserved (Sakai 2000; Sakai et al. 2002; Touchell and Dixon 2002).
In the vitrification method, a highly concentrated vitrification solution (PVS2) is used. This solution dehydrates tissues of explants sufficiently without causing injury so that they form a stable glass and thus remain alive, when they are plunged into LN. Key factors for successful cryopreservation are an increase in osmotic dehydration tolerance (osmotolerance) and a mitigation of the injurious effects of the shoot tip dehydration process (Sakai et al. 2002). Osmoprotection, treatment with a mixture of 2 M glycerol and 0.4 M sucrose (LS solution) following preculture with 0.3 M sucrose, significantly increased osmotolerance of shoot tips and improved the recovery growth (Matsumoto et al. 1994; Takagi et al. 1997; Charoensub et al. 1999; Hirai and Sakai 1999b; Lambardi et al. 2000; Pennycooke and Towill 2000, 2001). These positive effects of osmoprotection with or without preculture were also demonstrated in several tropical plants (Charoensub et al. 1999; Thinh et al. 1999; Kyesmu and Takagi 2000; Niino et al. 2000; Thinh and Takagi 2000).
Sweet potato is one of the most important root crops in tropical and sub-tropical countries and its genetic resources are conserved in field gene banks and/or in vitro culture. Cryopreservation of sweet potato using vitrification has been difficult because of its low tolerance to dehydration with PVS2 (Takagi et al. 1998) and its sensitivity to low temperature during cold-hardening. Recently, Pennycooke and Towill (2000, 2001) reported the successful cryopreservation of sweet potato using a combined vitrification-droplet method. In this method, shoot tips and one droplet of PVS2 were placed on a thin aluminum strip and immersed into partially solidified nitrogen (−208°C) to achieve rapid cooling (−400°C/min).
In the present study, we determined the optimal conditions for successful cryopreservation of sweet potato shoot tips using an encapsulation vitrification method without the use of any special technique and rapid cooling. The rate of recovery growth was about 80% for the three cultivars tested. This paper describes a simple, effective, encapsulation vitrification method for long-term conservation of sweet potato germplasm.
Materials and methods
Plant materials
In-vitro-grown sweet potato [Ipomoea batatas (L.) Lam.] 'Beni-azuma', 'Chikou-1gou' and 'Kogane-sengan' were used in this study. All in vitro plantlets were supplied by Dr. Kei Shimonishi, Kagoshima Biotechnology Institute (Kagoshima, Japan). Apical buds with one or two nodal segments (Fig. 1a) were excised from the plantlets and cultured on 20 ml basal MS (Murashige and Skoog 1962) medium supplemented with 30 g/l sucrose, 1 g/l casamino acids and 2 g/l Gellan-gum, in tissue culture dishes (90 mm ×20 mm) at 25°C under a 16 h photoperiod at 50 µmol m−2 s−1 for 14 days (stock culture). Media were adjusted to pH 5.7 prior to autoclaving at 121°C for 7 min. For micropropagation, nodal segments consisting of a piece of stem about 8 mm long were transferred to basal medium supplemented with 0.5 mg/l 6-benzyl aminopurine (BA) and incubated as above. Leaves were removed from nodal segments before their culture for the uniform growth of axillary buds (Fig. 1b). After 10–14 days incubation (Fig. 1c), shoot tips with three to four leaf primordia (about 1 mm long) were then excised for use in later experiments.

Stock cultures and micropropagation of sweet potato. a Apical buds with one or two nodal segments for initiating stock cultures. b Nodal segments without leaves cultured on basal medium supplemented with 0.5 mg/l 6-benzyl aminopurine (BA). c A nodal segment 14 days after transfer. Material: cv. Beni-azuma. Bars 10 mm
Encapsulation, preculture and osmoprotection
Excised shoot tips were suspended in a bead solution composed of Ca2+-free MS inorganic medium supplemented with 2% (w/v) Na-alginate (100–150 cp; Wako, Japan) and 30 g/l sucrose. A sterile 2 ml pipette was used to dispense a droplet of bead solution containing a single shoot tip into MS medium supplemented with 30 g/l sucrose and 0.1 M CaCl2 (encapsulation solution). Shoot tips were kept in the encapsulation solution for 30 min at 25°C to allow completion of the gelation process (to about 4 mm in diameter). Beads (10–15) were pre-incubated in 40 ml liquid basal MS medium supplemented with 30 g/l sucrose and 1 g/l casamino acid in a 100 ml Erlenmeyer flask for 24 h on a rotary shaker (90 rpm) before being transferred to a sucrose-enriched (0.3 M) liquid basal medium and precultured for 16 h. Pre-incubation and preculture were carried out under the same conditions as for stock culture. Osmoprotection was accomplished by incubating precultured beads in liquid MS medium supplemented with different concentrations (0.4–1.8 M) of sucrose with or without 2 M glycerol for 3 h on a rotary shaker (60 rpm) at 25°C.
Vitrification procedure
Between 10 and 15 encapsulated, precultured and osmoprotected shoot tips were dehydrated in 20 ml PVS2 in a 50 ml Erlenmeyer flask on a rotary shaker (60 rpm) at 25°C for various periods of time (0–100 min). PVS2 contains 30% (w/v) glycerol, 15% (w/v) ethylene glycol, 15% (w/v) DMSO and 0.4 M sucrose in MS medium (pH 5.8). After dehydration, beads were transferred to a 2 ml cryotube and suspended in 0.5 ml PVS2. Cryotubes were plunged directly into LN (cooling rate: about −200°C/min) and held there for at least 1 h.
Dilution process
After storing in LN, cryotubes were rapidly warmed in a water bath at 38°C for 2 min (rewarming rate: about 200°C/min). PVS2 was drained from the cryotubes and replaced twice at 10 min intervals with 1 ml 1.2 M sucrose solution (dilution solution).
Viability and plant regrowth
The LN-treated beads (Fig. 5a) were placed on recovery medium 1, consisting of basal medium supplemented with 0.5 mg/l BA and 1 mg/l gibberellic acid 3 (GA3) for 7 days and then transferred to recovery medium 2, consisting of basal medium supplemented with 0.5 mg/l GA3. The culture environment was the same as for stock cultures. Shoot formation was expressed as a percentage of the total number of shoot tips forming normal shoots 21 days after plating. Between 10 and 15 beads were treated in each of three replicates.
Results and discussion
In preliminary experiments, it was confirmed that both plantlets and excised shoot tips of sweet potato were too sensitive to tolerate cold-hardening for 14 days at 5°C, and dehydration with PVS2 for 3 h at 0°C. Thus, each step of the vitrification protocol—osmoprotection, dehydration with PVS2, and dilution—was carried out at 25°C.
The benefit of pre-incubating shoot tips before their preculture was examined using liquid MS medium containing 30 g/l sucrose for 24 h after encapsulation. As shown in Table 1, a 1-day pre-incubation produced a significantly higher level of recovery growth when followed by preculture with 0.3 M sucrose for 16 h. However, pre-incubation alone and preculture without pre-incubation gave very low recovery rates. Elimination of preculture in 0.3 M sucrose resulted in a decrease of recovery rate in wasabi (Matsumoto et al. 1994) and cassava (Charoensub et al. 1999), or in no survival at all in sweet potato (Pennycooke and Towill 2000). Thus, pre-incubation followed by preculture was an important step for the successful cryopreservation of sweet potato by encapsulation vitrification. These results appeared to agree with those of Pennycooke and Towill (2000, 2001). Longer periods of pre-incubation (>48 h) with 30 g/l sucrose and preculture (>24 h) with 0.3 M sucrose did not further improve the percentage of shoot formation (data not shown).
To determine the best combination of osmoprotectants for sweet potato shoot tips, solutions that included various concentrations of sucrose with or without 2 M glycerol were examined. As shown in Fig. 2, the highest percentage of shoot formation was obtained when precultured shoot tips were treated with 2 M glycerol and 1.6 M sucrose for 3 h at 25°C. Treatment with sucrose solution alone produced low percentages (below 20%) of shoot formation. Based on this, a mixture of 2 M glycerol plus 1.6 M sucrose was used as an osmoprotectant in subsequent experiments. The optimal incubation time in a mixture of 2 M glycerol and 1.6 M sucrose at 25°C was examined. As shown in Fig. 3, incubation for 3 h gave the highest percentage of shoot formation (about 80%).

Percentage of shoot formation for encapsulated vitrified shoot tips of sweet potato treated with various osmoprotectants. Encapsulated, pre-incubated and precultured shoot tips were treated with various osmoprotectants, including different concentrations of sucrose with or without 2 M glycerol for 60 min at 25°C before being dehydrated with a highly concentrated vitrification solution (PVS2) for 60 min at 25°C; 10–15 shoot tips were treated in each of three replicates. Material: cv. Beni-azuma. Bars standard error

Effect of incubation time in a mixture of 2 M glycerol and 1.6 M sucrose at 25°C on percentage of shoot formation of encapsulated sweet potato shoot tips cooled to −196°C. Encapsulated, pre-incubated and precultured shoot tips were osmoprotected with 2 M glycerol and 1.6 M sucrose for different lengths of time before being dehydrated with PVS2 for 60 min and plunged into liquid nitrogen (LN); 10–15 shoot tips were treated in each of three replicates. Material: cv. Beni-azuma. Bars standard error
The protective effect of osmoprotection might be due to osmotic dehydration, resulting in the concentration of cytosolic stress-responsive solutes that were accumulated during preculture with sucrose or sorbitol-enriched medium (Reinhoud 1996). During incubation in LS solution for 20 min, meristematic cells became plasmolyzed, producing concentrated spherical protoplasts (Matsumoto et al. 1998; Sakai et al. 2002). Plasmolysis might mitigate the mechanical stress incurred during severe dehydration (Hellergren and Li 1981; Tao et al. 1983; Jitsuyama et al. 1997). A higher concentration of sucrose (1.6 M) in LS solution and a longer period of osmoprotection were necessary to increase the osmotolerance of sweet potato shoot tips, although the actual protective action mechanism is poorly understood.
The pre-incubated, precultured and osmoprotected shoot tips were dehydrated with PVS2 at 25°C for various lengths of time to determine the optimal time of exposure to PVS2 before immersion in LN. The percentage of shoot formation of vitrified shoot tips increased considerably between 40 and 50 min and reached a maximum (about 70%) at 50–60 min. The shoot tips dehydrated with PVS2 up to 60 min but LN treatment (treated control) retained a high level of shoot formation (Fig. 4).

Effect of exposure time to PVS2 at 25°C on percentage of shoot formation of encapsulated sweet potato shoot tips cooled to −196°C. Encapsulated, pre-incubated and precultured shoot tips were osmoprotected with 2 M glycerol and 1.6 M sucrose for 3 h. They were then dehydrated with PVS2 for various lengths of time prior to a plunge into LN (+LN); 10–15 shoot tips were treated in each of three replicates. −LN, No LN treatment (no replicates). Material: cv. Beni-azuma. Bars standard error
Apices cryopreserved by vitrification with or without encapsulation have been reported to produce shoots directly without intermediary callus formation and genetic variants (Yamada et al. 1991; Fukai et al. 1991; Matsumoto et al. 1995a, 1995b; Touchell and Dixon 1995; Hirai et al. 1998; Hirai and Sakai 1999a, 1999b, 2001). Cryopreserved shoot tips of sweet potato were rapidly warmed, diluted and plated on recovery medium 1 for 7 days (Fig. 5a), then transferred to recovery medium 2. These shoot tips resumed growth and developed vigorous normal shoots (Fig. 5e, f, h, i). Roots developed within 14 days after placement of shoot tips on recovery medium 2 (Fig. 5i). These plants appeared identical to the non-treated phenotypes. However, cryopreserved shoot tips osmoprotected with 1.6 M sucrose alone did not develop into shoots and turned brown within 21 days (Fig. 5g).

Shoot formation from encapsulated sweet potato shoot tips cooled to −196°C. a Encapsulated vitrified shoot tips immediately after dilution and plating. b Encapsulated vitrified shoot tips 2 days after plating. c–e Encapsulated vitrified shoot tips 4, 7 and 10 days after plating, respectively. f Normal shoot (black arrow) 14 days after plating. Black arrowheads Swollen leaves. g Encapsulated vitrified shoot tips osmoprotected with 1.6 M sucrose alone. h, i Normal plantlets 18 and 21 days after plating, respectively. Black arrowheads Shoots. Material: cv. Beni-azuma. Bars 10 mm
This encapsulation vitrification protocol was tested with two other cultivars of sweet potato (Chikou-1gou and Kogane-sengan). The percentage of shoot formation of both of these cultivars exceeded 80% (Fig. 6).

Percentage of shoot formation of encapsulated shoot tips of three cultivars of sweet potato cooled to −196°C. Encapsulated, pre-incubated and precultured shoot tips were osmoprotected with 2 M glycerol and 1.6 M sucrose for 3 h before being dehydrated with PVS2 for 60 min, then plunged into LN; 10–15 shoot tips were treated in each of three replicates. Bars standard error
The encapsulation vitrification method has many advantages when compared to the vitrification method. The latter requires the handling of many small shoot tips floating or suspended in a solution. Their exposure to PVS2 must be precisely controlled. Thus, it is difficult to manipulate many small shoot tips in the solution at the same time. Encapsulated shoot tips are much easier to manipulate and permit greater flexibility in handling large amounts of material because the time scale at all steps is much longer than with the vitrification method. Encapsulated shoot tips probably undergo slower dehydration during exposure to PVS2. The encapsulation vitrification method has been successfully applied to cryopreservation of cell lines and somatic embryos of carrot (Hirai 2001).
The encapsulation vitrification method established in this study appears to be simple and effective for the cryopreservation of sweet potato germplasm. Further studies should be directed at examining the applicability of this technique across sweet potato genotypes.
Abbreviations
- BA :
-
6-Benzyl aminopurine
- DMSO :
-
Dimethyl sulfoxide
- GA 3 :
-
Gibberellic acid 3
- IAA :
-
Indole-3-acetic acid
- LN :
-
Liquid nitrogen
- MS :
-
Murashige-Skoog medium
- NAA :
-
1-Naphthaleneacetic acid
References
Bajaj YPS (1978) Tuberization in potato plants regenerated from freeze-preserved meristems. Crop Improv 5:137–141
Charoensub R, Phansiri S, Sakai A, Yongmanitchai W (1999) Cryopreservation of cassava in vitro-grown shoot tips cooled to −196°C by vitrification. Cryo-Lett 20:89–94
Fabre J, Dereuddre J (1990) Encapsulation-dehydration: a new approach to cryopreservation of Solanum shoot-tips. Cryo-Lett 11:413–426
Fukai S, Goi M, Tanaka M (1991) Cryopreservation of shoot tips of Chrysanthemum morifolium and related species native to Japan. Euphytica 54:201–204
Harding K, Benson EE (1994) A study of growth, flowering and tuberization in plants derived from cryopreserved shoot-tips of Solanum tuberosum. Cryo-Lett 15:59–66
Hellergren J, Li PH (1981) Survival of Solanum tuberosum suspension cultures to −14°C: the mode of action of proline. Physiol Plant 52:449–453
Hirai D (2001) Studies on cryopreservation of vegetatively propagated crops by encapsulation vitrification method. Rep Hokkaido Pref Agric Exp Stn 99:1–58
Hirai D, Sakai A (1999a) Cryopreservation of in vitro-grown axially shoot tip meristems of mint (Mentha spicata L.) by encapsulation vitrification. Plant Cell Rep 19:150–155
Hirai D, Sakai A (1999b) Cryopreservation of in vitro-grown meristems of potato (Solanum tuberosum L.) by encapsulation-vitrification. Potato Res 42:153–160
Hirai D, Sakai A (2001) Recovery growth of plants cryopreserved by encapsulation-vitrification. Bull Hokkaido Agric Exp Stn 80:55–64
Hirai D, Shirai K, Shirai S, Sakai A (1998) Cryopreservation of in vitro-grown meristems of strawberry (Fragaria × ananassa Duch.) by encapsulation-vitrification. Euphytica 101:109–115
Jitsuyama Y, Suzuki T, Harada T, Fujikawa S (1997) Ultrastructural study of mechanism of increased freezing tolerance to extracellular glucose in cabbage leaf cells. Cryo-Lett 18:33–44
Kyesmu PM, Takagi H (2000) Cryopreservation of shoot apices of yams (Dioscorea species) by vitrification. In: Engelmann F, Takagi H (eds) Cryopreservation of tropical plant germplasm. JIRCAS, Tsukuba, Japan, pp 411–413
Lambardi M, Fabbri A, Caccavale A (2000) Cryopreservation of white poplar (Populus alba L.) by vitrification of in vitro-grown shoot tips. Plant Cell Rep 19:213–218
Langis R, Schnabel-Preikstas BJ, Earle ED, Steponkus PL (1990) Cryopreservation of carnation shoot tips by vitrification. Cryobiology 27:657–658
Matsumoto T, Sakai S, Yamada K (1994) Cryopreservation of in vitro-grown apical meristems of wasabi (Wasabia japonica) by vitrification and subsequent high plant regeneration. Plant Cell Rep 13:442–446
Matsumoto T, Sakai A, Takahashi C, Yamada K (1995a) Cryopreservation of in vitro-grown apical meristems of wasabi (Wasabi japonica) by encapsulation-vitrification method. Cryo-Lett 16:189–206
Matsumoto T, Sakai A, Yamada K (1995b) Cryopreservation of in vitro-grown apical meristems of lily by vitrification. Plant Cell Tissue Organ Cult 41:237–241
Matsumoto T, Sakai A, Nako Y (1998) A novel preculturing for enhancing the survival of in vitro-grown meristems of wasabi (Wasabia japonica) cooled to −196°C by vitrification. Cryo-Lett 19:27–36
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue culture. Physiol Plant 15:473–497
Niino T, Seguel I, Murayama T (2000) Cryopreservation of vegetatively propagated species (mainly mulberry). In: Engelmann F, Takagi H (eds) Cryopreservation of tropical plant germplasm. JIRCAS, Tsukuba, Japan, pp 194–199
Pennycooke JC, Towill LE (2000) Cryopreservation of shoot tips from in vitro plants of sweet potato [Ipomoea batatas (L.) Lam.] by vitrification. Plant Cell Rep 19:733–737
Pennycooke JC, Towill LE (2001) Medium alternations improve regrowth of sweet potato (Ipomoea batatas [L.] Lam.) shoot tips cryopreserved by vitrification and encapsulation-dehydration. Cryo-Lett 22:381–389
Reinhoud PJ (1996) Cryopreservation of tobacco suspension cells by vitrification. Doctoral Paper, Rijks University, Leiden, pp 1–95
Sakai A (1995) Cryopreservation of germplasm of woody plants. In: Bajaj YPS (ed) Biotechnology in agriculture and forestry, vol 52. Springer, Berlin Heidelberg, New York, pp 53–69
Sakai A (2000) Development of cryopreservation techniques. In: Engelmann F, Takagi H (eds) Cryopreservation of tropical plant germplasm. JIRCAS, Tsukuba, Japan, pp 1–7
Sakai A, Kobayashi S, Oiyama I (1990) Cryopreservation of nucellar cells of navel orange (Citrus sinensis Osb. var. brasiliensis Tanaka) by vitrification. Plant Cell Rep 9:30–33
Sakai A, Matsumoto T, Hirai D, Charoensub R (2002) Survival of tropical apices cooled to −196°C by vitrification—development of a potential cryogenic protocol of tropical plants by vitrification. In: Li PH, Palva ET (eds) Plant cold hardiness—gene regulation and genetic engineering. Kluwer/Plenum, New York, pp 109–119
Takagi H, Thinh NT, Islam OM, Senboku T, Sakai A (1997) Cryopreservation of in vitro-grown shoot tips of taro (Colocasia esculenta (L.) Schott) by vitrification. 1. Investigation of basic conditions of the vitrification procedures. Plant Cell Rep 16:594–599
Takagi H, Thinh NT, Kyesm OM (1998) Cryopreservation of vegetatively propagated tropical crops by vitrification. Acta Hortic 461:485–495
Tao D, Li PH, Carter JV (1983) Role of cell wall in freezing tolerance of cultured potato cells and their protoplasts. Physiol Plant 58:527–532
Thinh NT, Takagi H (2000) Cryopreservation of in vitro-grown apical meristems of terrestrial orchids (Cymbidium spp) by vitrification. In: Engelmann F, Takagi H (eds) Cryopreservation of tropical plant germplasm. JIRCAS, Tsukuba, Japan, pp 441–443
Thinh NT, Takagi H, Yashima S (1999) Cryopreservation of in vitro-grown shoot tips of banana (Musa spp) by vitrification method. Cryo-Lett 20:163–174
Touchell DH, Dixon KW (1995) Cryopreservation for the conservation of native Australian endangered plants. In: Normah MN, Narimah MK, Clyde MM (eds) In-vitro conservation of plant genetic resources. University Kebangsaan, Malaysia, pp 169–180
Yamada T, Sakai A, Matsumura T, Higuchi S (1991) Cryopreservation of apical meristems of white clover (Trifolium repens L.) by vitrification. Plant Sci 78:81–87
Acknowledgement
The authors wish to thank Dr. Kei Shimonishi, Kagoshima Biotechnology Institute for supplying in-vitro-grown sweet potato plants
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by G.C. Phillips
Rights and permissions
About this article
Cite this article
Hirai, D., Sakai, A. Simplified cryopreservation of sweet potato [Ipomoea batatas (L.) Lam.] by optimizing conditions for osmoprotection. Plant Cell Rep 21, 961–966 (2003). https://doi.org/10.1007/s00299-003-0618-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00299-003-0618-5