
Abstract
The aims of the study were to determine prognostic factors for survival and causes of death in a cohort of patients with systemic sclerosis (SSc). This was a cohort study of SSc patients in single rheumatologic center from January 1998 to August 2012. They fulfilled the American College of Rheumatology classification criteria for SSc or had calcinosis Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia or sine sclerosis. Causes of death were classified as SSc related and non-SSc related. Kaplan–Meier and Cox proportional hazard regression models were used in univariate and multivariate analysis to analyse survival in subgroups and determine prognostic factors of survival. The study includes 220 patients (192 female, 28 male). Out of thirty-two (14.5 %) who died, seventeen (53.1 %) deaths were SSc related and in nine (28.1 %) non-SSc-related causes, and in six (18.8 %) of patients causes of death were not defined. Overall survival rate was 92.6 % (95 % CI 87.5–95.7 %) after 5 years and 82.3 % (95 % CI 73.4–88.4 %) after 10 years. Pulmonary involvement was a major SSc-related cause of death, occurred in seven (41.1 %) patients. Cardiovascular events were leading cause of in overall death (11) 34.3 % and 6 in non-SSc-related death. Independent risk factors for mortality were age >50 at diagnosis (HR 5.10) advance pulmonary fibrosis (HR 11.5), tendon friction rub at entry (HR 6.39), arthritis (HR 3.56). In this first Middle Eastern series of SSc registry, pulmonary and cardiac involvements were the leading cause of SSc-related death.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The natural history of systemic sclerosis (SSc) varies in different cases and depends on demographic and severity of organ involvement. Previous studies indicated that demographic factors such as male gender [1–7], older age at disease presentation [1, 2, 8–10] and black race [1] are associated with poor survival rate.
Moreover, diffuse and greater extension of skin involvement [4, 6, 11–13] as well as internal organs including lung [4, 6], heart (pericarditis, clinical right side heart failure) [6, 12, 14–18], kidney [6–8, 13, 14], gastrointestinal tract [19] and joint [20] contracture are all associated with poor prognosis. Markers such as anti-topoisomerase antibody [16], anemia [13, 21], increased erythrocyte sedimentation rate (ESR) [6, 8, 13] and C-reactive protein (CRP) level [22], proteinurea without scleroderma, renal crisis and abnormal urine sedimentation are also showed to be associated with poor prognosis [23, 24].
Disease prognosis in SSc is highly variable. Different studies have reported different survival rates. Five-year survival rates are reported as 77, 85 and 90 %. The 10-year survival rates have also been variably reported as 59, 77 and 82 % [24–26].
It is important to note that the reported survival rates in clinical studies on SSc patients may encounter selection bias (4). Survival rate analysis could be affected by selection of the time of the first symptom, the first diagnosis or the entry time into the study. As an example in retrospective cohort study of scleroderma in which the time of first onset of symptom (Raynaud) is selected as starting point of a study, instead of the time of diagnosis (a result in time interval difference of ≥5 years or more), no mortality occurred as predicted and 5-year survival may even be better than general population. In cohort studies that starting point could not be precisely selected, immortality bias may arise. In addition, loss of follow-up with incomplete ascertainment of vital status could be another reason that would result in survival rate reports’ variability.
Variances in prognostic factors in survival studies, however, may result from differences in statistical analysis and applied models such as univariate Kaplan–Meier or multivariate Cox proportional hazards analysis.
Our cohort is unique for two reasons: Firstly, it represents mortality and survival data in relatively large population of scleroderma patients in the Middle East region; secondly, we used the data collected from the very first locally established systemic sclerosis registry in Iran.
Methods
The CRIS database
Patients’ data were obtained from Scleroderma Registry which is part of Clinical and Research Rheumatology Information System (CRIS). This is a single-center electronic database which is designed locally for prospective cohort study and is used to record information on patient demographic, disease progression and laboratory findings. In our center in clinic of rheumatology, referred patients are initially visited by a rheumatologist. If their diagnosis is confirmed, after obtaining an informed written consent, the physician takes the patients' complete history and performs appropriate physical examination and paraclinical data are then saved for follow-up visits.
Data related to six groups of rheumatic disease including SSc, rheumatoid arthritis, seronegative arthropathy, inflammatory muscle disease, systemic lupus and vasculitis are recorded in CRIS. The registry has been in use since February 2008 when we initiated an open cohort study on SSc. Data for patients who visited before February 2008 were transferred from paper-based records to CRIS databank. Additional data that were registered in the scleroderma registry include onset of symptoms, date of diagnosis, date of first visit in the clinic, demographic, clinical and paraclinical findings at the baseline and throughout the study. All radiographic images are saved for prospective follow-ups. Frequency of clinical features, disease outcome in regard to other organs and systemic involvements, cumulative survival data across patients groups with specific antibody are also recorded in CRIS. In the scleroderma registry, we specified subtypes of SSc such as limited cutaneous (lcSSc), diffuse cutaneous (dcSSc) [27], overlap syndrome [28] and sine scleroderma [29, 30].
Inclusion and exclusion criteria
From 247 patients who were visited at Firoozgar Hospital, from January 1998 to August 2012, and followed until August 2013, we included 220 patients who had fulfilled the inclusion criteria.
Of those, 207 patients fulfilled the American College of Rheumatology classification [31] or had calcinosis Raynaud’s phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia [27], and 13 patients diagnosed sine scleroderma according to established criteria [29, 30] and categorized under limited SSc group as suggested in other study. [29, 30]. Patients with skin involvement were subdivided into two subtypes of lcSSc and dcSSc according to LeRoy et al. [27]. Endpoint of study was time of death or time to make a phone call or last visit for patients who survived.
We exclude 27 patients, 9 patients who were diagnosed as early Scleroderma or overlap and 18 (7.2 %) patients who had completely missed follow-up visits, and we lost contact with them.
The disease onset was defined as the time when Raynaud’s phenomenon or non-Raynaud’s scleroderma-related sign or symptoms were noticed by patients. The time that a patient was first diagnosed recorded as time of diagnosis, even in those patients who were diagnosed months or years before their first visit. In order to do the survival analysis, the first time the diagnosis is performed is considered as the starting point. Patients who did not come back for their follow-up visits after 12 months were contacted via phone to check on their well-being.
Survival prognostic factors and causes of death
In addition to demographic and serological factors, visceral involvements have been also depicted as prognostic factor and cause of death in the previous studies [5, 6, 8, 32]. In this study, we followed SSc-related cause of death based on visceral organ involvement according to Steen and Medsger [32] with some modifications;
-
1.
Advanced Pulmonary fibrosis determined by demonstration of more than 20 % fibrosis on high-resolution computerized tomography (HRCT) of chest and/or restrictive pattern on pulmonary function test that is forced vital capacity (FVC) of lower than 70 % of predicted value or <60 % of predicted carbon monoxide diffusing capacity (DLCO).
-
2.
Pulmonary hypertension (PH); Right heart catheterization was not performed on all patients; therefore, we did not consider pulmonary arterial hypertension (PAH) in our series. But in this study, we assessed relation of pulmonary arterial pressure (PAP) >50 mmHg in echocardiography with survival as a prognostic factor.
-
3.
Cardiac involvement includes pericardial effusion as per echocardiographic evidence or HRCT, left ventricular systolic ejection fraction <55 % by echocardiography (Echo) and conduction disease, or arrythmia.
Clinical heart failure and left ventricular failure were defined as ejection fraction of <50 % in echocardiography (in the absence of systemic or pulmonary hypertension and without significant renal involvement or nodal or ventricular arrhythmia) and conduction defects (not attributable to other cardiac conditions) requiring treatment. All of the above were considered as cardiac prognostic factor or cause of death. In addition, we considered smoking and hypertension as attributing causes of death.
-
4.
Renal involvement was defined as scleroderma renal crisis (SRC) and renal abnormalities independent of renal crisis (proteinuria, hematuria, cellular cast, BUN >25, or creatinine >1.2 mg/dl) mild isolated hypertension requiring medication (but without typical renal crisis) during the course of the disease. Scleroderma renal crisis (SRC) was defined as malignant hypertension and/or rapidly progressive renal insufficiency and/or microangiopathic hemolytic anemia.
-
5.
Gastrointestinal involvement was defined as gastrointestinal reflux, esophageal hypo motility that is diagnosed either by manometery, esophageal dilation on HRCT or cineradiographic. Small bowel involvement defined as diarrhea due to bacterial overgrowth that needed antibiotics. Body mass index ≤18.5 kg/m2 is considered as an index of malnutrition.
-
6.
Multiorgan failure is defined as severe simultaneous damage from >1 SSc-related organ involvement for which it was difficult to determine the primary organ failure that was the cause of death, for example patients with interstitial lung disease who also presented PH in the follow-up were considered into both categories causing death.
A patient is considered to have non-SSc-related death if they had died of infection, cancer, stroke, myocardial infarction, sudden death without prior scleroderma heart disease. If infection occurs in setting of scleroderma-related organ failure (severe PAH, right heart failure or renal crisis), we assigned cause of death as SSc related.
In the event of a death report in our center, hospital records were reviewed to document the date and cause of death. If death occurred outside our center, information on the time and cause of death was obtained by following up phone calls to the family or treating physician.
Cause of death was determined in 21 patients. In 5 patients, review of patient’s history and family description of the events around the death led us to determining the cause of death. However, neither if the above method could help defining the cause of death in 6 of our patients.
Serology study
Sera of all patients visited from 1998 to 2013 were collected and stored at −80 °C until use. For analysing the pattern of ANA in sera, indirect immunofluorescence technique was used via Mosaic HEp-20-10 Liver (monkey), (EUROIMMUN, Lübeck Germany). In order to detect anti-centromere, Anti-TOPO I, Enzyme link Immunosorbent Assay (ELISA) Orgentec kit was used according to the manufacturer’s instructions. Absorbances were read at 450 nm on an ELISA plate reader.
Statistical analysis
The statistical methods that were used in univariate analysis all demographic and clinical characteristic of SSc patients assessed by univariate Cox regression model, the variables with p ≤ 0.2 in univariate analysis and prognostic factors suggested by other studies were conducted by backward stepwise multivariate Cox-proportional hazards regression model with entry and removal probabilities equal to 0.05 and 0.1, respectively. The effect of all demographic and clinical variables on survival of SSc patients was assessed by univariate Cox regression model. The variables with p<= 0.2 in univariate analysis as well as prognostic factors suggested by other studies were included in backward stepwise multivariate Cox proportional hazard regression model (with entry and removal probabilities set at 0.05 and 0.1, respectively) to identify multiple predictors of overall survival. Proportional hazards assumption was evaluated using the Chi square test of correlation coefficient between transformed survival time and scaled Schoenfeld residuals. Survival curves were estimated by Kaplan–Meier method and were compared between different subgroups using log-rank test.
Results
Characteristic of samples
The study includes 220 patients (192 females, 28 males). Female-to-male ratio was 6.9 to 1. Mean age at onset of disease was 36.2 (SD = 13.1). Mean age of all patients at the time of diagnosis was (mean ± SD) 38.9 ± 13.2, and mortality rate was higher in patients who were older at the time of diagnosis 47.9 ± 13.1 than those who survived 37.4 ± 12.6 (p = 0.001). Forty-five patients were ≥50 years old at the time of diagnosis. Overall, 20 patients (62.5 %) who died and 46 (24.5 %) of those who survived were ≥50 years old.
A total of 119 (54.0 %) patient presented limited cutaneous subtype, 88 (40 %) were of diffuse cutaneous subtype and 13 (5.9 %) were of sine scleroderma subtype of SSc. We bundled the two sine scleroderma and limited SSc groups as one group under limited SSc subtype.
Time interval from onset of first symptom to diagnosis of SSc was (mean ± SD) 2.9 ± 4.3 years. This interval was 4.1 ± 5.5 years in deceased and 2.7 ± 4.0 in survived patients (p = 0.17). At the time of analysis, the mean disease follow-up time until death or censored at last visit or last call was 6.6 ± 5.5 years. Mean disease follow-up in those who deceased and survived were 8.0 ± 6.8 and 6.4 ± 5.5, respectively, with no significant difference.
Total follow-up time was 1456 person-years. Patients in our study had a wide range of disease duration from a minimum of 12 months to maximum of 27.9 years (Range 26.9 years). The baseline and clinical characteristics and paraclinical data are shown in Table 1.
Causes of death and survival rate
A total of 32 (14.5 %) patients died during follow-up. The primary causes of death in 17 patients (53.1 %) who died were SSc related. From those, lung involvement was leading cause of death in 7, cardiomyopathy in 4 patients, multi organ involvement in 3, SRC in 2 and gastrointestinal bleeding due to watermelon stomach in 1 patient.
Non-scleroderma-related causes of death were found in 9 of died patients. Of those, 6 patients die due to cardiovascular event, 2 due to malignancy (1 esophageal adenoma and 1 patient because of metastatic bone cancer of unknown origin) and 1 death was related to infection. Cause of death in 6 other patients (18.8 %) was not found (Table 2).
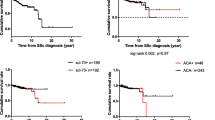
Kaplan–Meier analysis showed a survival rate of 92.6 % (95 % CI 87.5–95.7 %) after 5 years and 82.3 % (95 %CI 73.4–88.4 %) after 10 years. Survival rate after 5, 10 years in diffuse was 91.3 % (95 % CI 81.4–96.0), 79.9 % (95 % CI 64.3–89.2) and in the limited 93.4 % (95 % CI 86.6–96.8), 83.7 % (95 % CI 71.7–90.9) subtypes of disease was not differed (p = 0.71) (Fig. 1).

Pairs of survival curve for 220 patients with systemic sclerosis, by subgroups showed. Horizontal axis represents duration from the diagnosis of SSc, and the number of events was shown. Survival rate after 5, 10, 15, 20 years in diffuse and limited subset of disease was not differed (p = 0.71)
Univariate analysis
Analyses by univariate Cox analysis method are showed in Table 3.
Analysis revealed that the patients with age of over 50 at time of diagnosis, tendon friction rub, FVC <70 %, DLCO <60 %, advance pulmonary hypertension and pulmonary arterial pressure in echocardiography (PAP) >50 mmHg associated with poor outcome (p value ≤0.05). In Fig. 2, the Kaplan–Meier survival curves are shown for parameters with significant impact on dying in univariate analysis.

Kaplan–Meier graph for survival of systemic sclerosis (SSc) on 220 patients of whom 32 died. Horizontal axis represents duration from the diagnosis. Better survival was seen in patients with a age lower than 50 (p = 0.001), b patients without friction rub at entry (p = 0.02). c Patient without pulmonary fibrosis or mild fibrosis (p = 0.001) and d *patients without Pulmonary arterial hypertension >50 in echocardiography (p = 0.02). Graph for FVC and DLCO was not shown
Moreover, other variables including gender, calcinosis, arthritis, pericardial effusion, scleroderma renal crisis which in univariate analysis had p values ≤0.2 were included in the multivariate Cox proportional hazard model. The parameters with p > 0.20 that had no significant impact on dying were not considered in multivariate analysis, including the time interval from the first symptom (Raynaud, or non-Raynaud) to diagnosis, disease subtypes, smoking status, vascular manifestations [Raynaud seen by physician, telangiectasia (hands/face/lips), digital pitting ulcer, digital ulcer/gangrene/amputation body mass index (BMI) <18.5, ESR, hemoglobin and autoantibodies (ACA, TOPO, ANA)].
Multivariate analysis
When using Cox-proportional hazard model, we observed that Age > 50 years at time of diagnosis, friction rub at entry, arthritis and advanced pulmonary fibrosis were significant contributor factors in worsening prognosis. In our study, we could conclude these factors as independent prognostic factors for poor survival (Table 4).
Discussion
Baseline characteristic
Compared to previous report from European and American studies and published figures of scleroderma patients, patients included in this cohort had some baseline differences in regard to clinical features and serology, also earlier disease onset and diagnosis. Our patients were younger at the disease onset compared to a Hungarian study in which mean age at the diagnosis was 44 (range 33–53) years, and in a separate meta-analysis [33] 44.4 ± 3 years in the limited group and 47.8 ± 4.3 years in the diffuse group of patients [8]. However, a Canadian study reported older age range at the disease onset in their study population, 50.7 ± 2.3 years in deceased and 47.8 ± 1.3 years in survived patients [11].
In contrast to other studies [5, 8, 10], anti-Topo abs were the most prevalent antibodies in our series, and there was low prevalence of malignancy and scleroderma renal crisis. These differences may be attributed to the genetic and geographic background.
Survival and causes of death
In this cohort, cumulative 5- and 10-year survivals from diagnosis were 93, 83 % with a mortality rate of 14.5 %. This is consistent with studies that had shown improvement of SSc survival in the last decade [8, 9, 11, 26, 32, 34, 35]. Survival rates varied in different studies. Overall, 5- and 10-year survivals have been reported as 81, 71 % in Danish [35], 85, 72 % in Hungarian [8], 90, 84 % in Brazilian [5], 90, 82 % in Canadian [13] and 85.4, 80 % in Korean [9] studies.
The survival rates in previous studies depended on the selected age at disease onset, age at diagnosis or age at the study entry. As well as disease stage at time of entry (early vs late), ethnicity, number of patients with diffuse subtype of disease, severity of organ involvement and country-specific healthcare policies.
A total of 32 deaths have been occured in our study population. Seventeen deaths were considered SSc related and 9 were non-SSc related. Similar to other studies, pulmonary [16, 32, 34] and cardiac [3, 5, 6] involvements are the most common causes of death. Our finding is consistent with the previous studies in that cardiovascular diseases also represent the major cause of non-SSc-related death [34, 35].
In our cohort, cardiovascular (CV) events caused 31.2 % of overall mortalities. In patients with SSc disease, the UK study showed 30 % of deaths [36] and in a Swedish study 20 % of deaths were due to CV events [4]. These findings may support the previously described association between SSc and macrovascular disease [35, 37].
In this cohort, scleroderma renal crisis represent the cause of death in 6.4 % of all deaths which is also consistent with other studies in Asia and the US [9, 32, 38]. The SRC was rare in Iranian SSc patients with event rate of 1.8 %, and its prevalence was lower than reported rates from Korean 2.7 %, [9] Japanese 2.8 % [38] and EUSTAR cohort 2.3 % [39].
Malignancy was the cause of death in 6.4 % of patients and had occured during first year after disease diagnosis. None of the patients who developed cancer had received cyclophosphamide before diagnosis of malignancy. In the two patients who died because of cancer, anti-Topo I antibodies were positive and had diffuse subtype of the disease. The incidence of malignancy in SSc patients have been reported from 3.6 % in Pitsburgh to 10.7 % in south Australian studies [40]. A Danish population-based cohort showed that SSc increases the risk of smoking and alcohol-related cancer [41]. Additionally, male gender in SSc patients was a risk factor for cancer [41].
We did not find a consistency between the organ involvements at the time of diagnosis with organ system involvement in the course of the disease that ultimately could be a cause of death.
Predictors of death in SSc
Results of our cohort in Cox proportional hazards mode were consistent with the reports from previous studies that have delineated the pulmonary fibrosis, [34] elderly age [1, 2, 8, 9], friction rub [42], which were among the independent predictors of mortality. We found survival rate correlated with the age; patients who developed the disease and diagnosed at younger age had better survival than those who died. In other words, age over 50 year was an independent risk factor of death.
The current study demonstrated no relation with mortality and the time interval between symptoms to diagnosis and disease subtypes. We speculate this may be due to death occurring in patients with severe diffuse before their referral to the clinic.
In our study, arthritis was an independent predictive factor of mortality. It has been supposed that arthritis might be a predictive factor of the diffuse scleroderma and may carry with poor prognosis. This issue is under investigation in EUSTAR database [43].
We did not find FVC <70 % and DLCO <60 % as independent mortality risk factor which could indicate that advanced pulmonary disease was a better predictor of death than each of the above-mentioned factors.
We checked autoantibodies in 91.4 % of patients in cohort study whose patients entered the study since many years ago; presence of missing data is inevitable as depicted in Italian survival study in one-quarter of patients autoantibodies were not evaluated [6].
According to population studied, there is a variation prevalence of ANA across different population. ANA are reported in range of 84 % in Sweden study [4] to 98.7 % of patients in EULAR scleroderma trial and Research group database [39].
It was previously shown that anti-Topoisomerase I antibodies are an independent survival prognostic factor [3, 11, 16] But we did not find such association which might be due to high prevalence of anti-Topo abs and low prevalence of ACA abs in sera of patients in this cohort. Low prevalence of ACA abs in our study is consistent with the results from our own previous study [44] and other Asian study [9, 45].
Among 230 SSc patients included in a Korean study (56 % had lcSSc, 43.9 % dcSSc form of disease), ACA was detected in sera of 31 (13.6 %) and anti-Topo in 118 (53.6 %) patients [9]. Similarly, 31 Malaysian ACA was detected in 5 (16.1 %) patients [45]. Our center is running studies to evaluate the role of genetic background on prevalence of autoanibodies in Iranian patients.
In our study, among ACA-positive SSc patients, there were 14 patients (93.8 %) with lcSSc and (6.2 %) dcSSc subtype of disease anti-Topo abs were found in 82.1 % dcSSc patients and 60.9 % patients with lcSSc. Although this finding may disconcord with studies that supposed anti-Topo and ACA abs are classically associated with diffuse and limited subtype of disease [4, 39], but our results concordant with study in Iran [44] and recent Chinese cohort that ACA was found in 55.4 % patients with dcSSc form of disease and in Malaysian study that anti-TOPO abs is found in 50 % of lcSSc patients [45, 46].
Limitations
Current study had some limitations in determining causes of death. Our study included patients from single center and therefore cannot represent all patients in our country; the short number of deaths and similar survival in dcSSC an lcSSC patients may be due to selection bias in single center study. This is the first study of its kind in Iran and Middle East region and we hope that this study could be looked at future investigators. We were not able to properly evaluate pulmonary arterial hypertension (PAH). This was due to right heart catheterization, which was not conducted as a part of diagnosis of PH in the patients with high PAP in echocardiography.
As referred to in other literatures, causes of death are hard to retrospectively be confirmed. To determine causes of death in 4 patients, we considered the family description of the events surrounding the death and then recorded data for severity of organ involvement [34, 47].
In cases with more certain causes of death, the bias in coding the death certification could occur due to chronic known disease and that none of our patients were submitted to autopsy. This might contribute to a low incidence of infection and sepsis in the setting of severe organ failure and not being considered as causes of death.
It is sometimes impossible to estimate the exact relation of death to SSc, for example in ischemic heart disease cases; SSc disease itself is a contributing factor to microvascular [48, 49] and macrovascular changes [4, 35, 37] that might aggravate the problem.
The mean disease duration from first time of diagnosis to last visit or death was 6.6 ± 5.5 years. Our conclusion may change over time as we follow survived patients for longer duration. The SMR the most reliable mortality rate was not estimated in this study.
In summary, results from analysis of the data from this cohort demonstrated cardiac and pulmonary involvement (pulmonary fibrosis or pulmonary arterial pressure >50 mmHg) which were the leading causes of SSc-related death. Among clinical and immunological variables, age, tendon friction rub, advances pulmonary and arthritis were independently associated with poorer prognosis in patients with SSc.
References
Medsger TA Jr, Masi AT, Rodnan GP, Benedek TG, Robinson H (1971) Survival with systemic sclerosis (scleroderma). A life-table analysis of clinical and demographic factors in 309 patients. Ann Intern Med 75:369–376
Perez-Bocanegra C, Solans-Laque R, Simeon-Aznar CP et al (2010) Age-related survival and clinical features in systemic sclerosis patients older or younger than 65 at diagnosis. Rheumatology 49:1112–1117
Fransen J, Popa-Diaconu D, Hesselstrand R et al (2011) Clinical prediction of 5-year survival in systemic sclerosis: validation of a simple prognostic model in EUSTAR centres. Ann Rheum Dis 70:1788–1792
Hesselstrand R, Scheja A, Akesson A (1998) Mortality and causes of death in a Swedish series of systemic sclerosis patients. Ann Rheum Dis 57:682–686
Sampaio-Barros PD, Bortoluzzo AB, Marangoni RG et al (2012) Survival, causes of death, and prognostic factors in systemic sclerosis: analysis of 947 Brazilian patients. J Rheumatol 39:10
Ferri C, Valentini G, Cozzi F et al (2002) Systemic sclerosis: demographic, clinical, and serologic features and survival in 1,012 Italian patients. Medicine 81:139–153
Lee P, Langevitz P, Alderdice CA et al (1992) Mortality in systemic sclerosis (scleroderma). Q J Med 82:139–148
Czirják L, Kumánovics G, Varjú C et al (2008) Survival and causes of death in 366 Hungarian patients with systemic sclerosis. Ann Rheum Dis 67:59–63
Kim J, Park SK, Moon KW, Lee EY, Lee YJ, Song YW et al (2010) The prognostic factors of systemic sclerosis for survival among Koreans. Clin Rheumatol 29:297–302
Simeón CP, Armadans L, Fonollosa V, Solans R, Selva A, Villar M et al (2003) Mortality and prognostic factors in Spanish patients with systemic sclerosis. Rheumatology 42:71–75
Al-Dhaher FF, Pope JE, Ouimet JM (2010) Determinants of morbidity and mortality of systemic sclerosis in Canada. Semin Arthritis Rheum 39:269–277
Steen VD, Medsger TA Jr (2001) Improvement in skin thickening in systemic sclerosis associated with improved survival. Arthritis Rheum 44:2828–2835
Scussel-Lonzetti L, Joyal F, Raynauld JP, Roussin A, Rich E, Goulet JR et al (2002) Predicting mortality in systemic sclerosis: analysis of a cohort of 309 French Canadian patients with emphasis on features at diagnosis as predictive factors for survival. Medicine 81:154–167
Ruangjutipotan S, Kasitanon N, Louthrenoo W, Sukitawut W, Wichainun R (2002) Causes of death and poor survival prognostic factors in Thai patients with systemic sclerosis. J Med Assoc Thai 85:1204–1209
Jacobsen S, Ullman S, Shen GQ, Wiik A, Halberg P (2001) Influence of clinical features, serum antinuclear antibodies, and lung function on survival of patients with systemic sclerosis. J Rheumatol 28:2454–2459
Ioannidis JP, Vlachoyiannopoulos PG, Haidich AB, Medsger TA Jr, Lucas M, Michet CJ et al (2005) Mortality in systemic sclerosis: an international meta-analysis of individual patient data. Am J Med 118:2–10
Arias-Nuñez MC, Llorca J, Vazquez-Rodrigues TR, Gomez-Acebo I, Miranda-Filloy JA, Martin J et al (2008) Systemic sclerosis in northwestern Spain: a 19-year epidemiologic study. Medicine 87:272–280
Martini G, Vittadello F, Kasapçopur O, Magni Manzoni S, Corona F, Duarte-Salazar C et al (2009) Factors affecting survival in juvenile systemic sclerosis. Rheumatology 48:119–122
Mayes MD, Lacey JV, Beebe-Dimmer J, Gillespie BW, Cooper B, Laing TJ et al (2003) Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum 48:2246–2255
Clements PJ, Hurwitz EL, Wong WK, Seibold JR, Mayes M, White B et al (2000) Skin thickness score as a predictor and correlate of outcome in systemic sclerosis. Arthritis Rheum 43:2445–2454
Altman RD, Medsger TA Jr, Bloch DA, Michel BA (1991) Predictors of survival in systemic sclerosis (scleroderma). Arthritis Rheum 34:403–413
Nagy Z, Czirják L (2005) Increased C reactive protein and aminoterminal propeptide of type III procollagen levels are unfavourable predictors of survival in systemic sclerosis (scleroderma). Clin Exp Rheumatol 23:165–172
Bulpitt KJ, Clements PL, Lachenbruch PA, Paulus HE, Peter JB, Agopian MS et al (1993) Early undifferentiated connective tissue disease: III. Outcome and prognostic indicators in early scleroderma (systemic sclerosis). Ann Intern Med 118:602–609
Bryan C, Knight C, Black CM, Silman AJ (1999) Prediction of five-year survival following presentation with scleroderma development of a simple model using three disease factors at first visit. Arthritis Rheum 42:2660–2665
Tuffanelli DL, Winkelmann RK (1961) Systemic scleroderma, a clinical study of 727 cases. Arch Dermatol 84:359–371
Joven BE, Almodovar R, Carmona L, Carreira PE (2010) Survival, causes of death, and risk factors associated with mortality in Spanish systemic sclerosis patients: results from a single university hospital. Semin Arthritis Rheum 39:285–293
LeRoy EC, Black C, Fleischmajer R et al (1988) Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol 15:202–205
Bennett RM (1990) Scleroderma overlaps syndromes. Rheum Dis Clin North Am 16:185–198
Poormoghim H, Lucas M, Fertig N, Medsger TA Jr (2000) Systemic sclerosis sine scleroderma: demographic, clinical, and serologic features and survival in forty-eight patients. Arthritis Rheum 43:444–451
LeRoy EC, Medsger TA Jr (2001) Criteria for the classification of early systemic sclerosis. J Rheumatol 28:1573–1576
Preliminary criteria for the classification of systemic sclerosis (scleroderma) (1980) Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum 23:581–590
Steen VD, Medsger TA (2007) Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis 66:940–944
Rubio-Rivas M, Royo C, Simeón CP, Corbella X, Fonollosa V (2014) Mortality and survival in systemic sclerosis: systematic review and meta-analysis. Semin Arthritis Rheum 44:208–219
Tyndall AJ, Bannert B, Vonk M et al (2010) Causes and risk factors for death in systemic sclerosis: a studyfrom the EULAR Scleroderma Trials and Research (EUSTAR). Ann Rheum Dis 69:1809–1815
Jacobsen S, Halberg P, Ullman S (1998) Mortality and causes of death of 344 Danish patients with systemic sclerosis (scleroderma). Br J Rheumatol 37:750–755
Bryan C, Howard Y, Brennan P, Black C, Silman A (1996) Survival following the onset of scleroderma: results from a retrospective inception cohort study of the UK patient population. Br J Rheumatol 35:1122–1126
Belch JJ, McSwiggan S, Lau C (2008) Macrovascular disease in systemic sclerosis: the tip of an iceberg? Rheumatology 47(Suppl 5):v16–v17
Hashimoto A, Tejima S, Tono T et al (2011) Predictors of survival and causes of death in Japanese patients with systemic sclerosis. J Rheumatol 38:1931–1939
Walker UA, Tyndall A, Czirják L et al (2007) Clinical risk assessment of organ manifestations in systemic sclerosis: a report from the EULAR Scleroderma Trials and Research group database. Ann Rheum Dis 66:754–763
Wooten M (2008) Systemic sclerosis and malignancy: a review of the literature. South Med J 101:59–62
Olesen AB, Svaerke C, Farkas DK, Sørensen HT (2010) Systemic sclerosis and the risk of cancer: a nationwide population-based cohort study. Br J Dermatol 163:800–806
Doré A, Lucas M, Dana Ivanco D, Medsger TA, Domsic R (2013) The significance of palpable tendon friction rubs in early diffuse cutaneous systemic sclerosis. Arthritis Care Res 65:1385–1389
Avouac J, Clements PJ, Khanna D et al (2012) Articular involvement in systemic sclerosis. Rheumatology 51:1347–1356
Poormoghim H, Moghadam AS, Moradi-Lakeh M et al (2013) Systemic sclerosis: demographic, clinical and serological features in 100 Iranian patients. Rheumatol Int 33:1943–1950
Sujau I, Ng CT, Sthaneshwar P et al (2015) Clinical and autoantibody profile in systemic sclerosis: baseline characteristics from a West Malaysian cohort. Int J Rheum Dis 18:459–465
Wang J, Assassi S, Guo G et al (2013) Clinical and serological features of systemic sclerosis in a Chinese cohort. Clin Rheumatol 32:617–621
Steen VD, Medsger TA Jr (2000) Severe organ involvement in systemic sclerosis with diffuse scleroderma. Arthritis Rheum 43:2437–2444
Kahan A, Nitenberg A, Foult JM et al (1985) Decreased coronary reserve in primary scleroderma myocardial disease. Arthritis Rheum 28:637–646
Champion HC (2008) The heart in scleroderma. Rheum Dis Clin North Am 34:181–188
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors declare that they have no conflict of interest.
Ethical standard
After obtaining an informed written consent, patients data have entered Scleroderma Registry, Clinical and Research Rheumatology Information System (CRIS), which is allowed by the Medical Research Ethics Committee of the Iran University Medical Sciences, for clinical study (registration code: 76, No. P/900789).
Rights and permissions
About this article

Cite this article
Poormoghim, H., Andalib, E., Jalali, A. et al. Survival and causes of death in systemic sclerosis patients: a single center registry report from Iran. Rheumatol Int 36, 925–934 (2016). https://doi.org/10.1007/s00296-016-3475-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-016-3475-6