
Abstract
Osteonecrosis is commonly present in patients with antiphospholipid syndrome (APS) and systemic lupus erythematosus (SLE). Treatment of this condition remains extremely controversial. We present a treatment strategy of avascular necrosis of the knee in a patient with catastrophic antiphospholipid syndrome with a history of SLE and APS. Aggressive treatment with 12 rounds of plasmapheresis, intravenous immunoglobulin, rituximab, and cylophosphamide led to the patient’s recovery with no recurrence of symptoms during 16 months of follow up. In this report, we further discuss the pathogenesis of osteonecrosis and current understanding of the treatment of this disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Antiphospholipid syndrome (APS) is a thrombotic autoimmune disorder that has been described to be associated with high levels of antiphospholipid antibodies, including lupus anticoagulant antibodies, anti-cardiolipin antibodies, and β2-glycoprotein I antibodies [1]. APS is often found in the presence of other autoimmune diseases, such as systemic lupus erythematosus (SLE), in which case the disease is termed “secondary APS”. Though APS is a well-documented entity, certain manifestations of the syndrome such as osteonecrosis due to thrombotic events in patients with secondary APS and SLE have been underappreciated. There are several important risk factors for the development of osteonecrosis in patients with APS and SLE including glucocorticoid use in the treatment of SLE and the presence of antiphospholipid antibodies [1]. There is currently no standardized treatment for osteonecrosis perhaps because the pathogenesis of osteonecrosis itself remains controversial and unclear; traditional pharmacological approaches including anticoagulants, antihyperlipidemic drugs, and bisphosphonates have yielded no concrete results [2]. We present a case of right knee osteonecrosis secondary to catastrophic antiphospholipid syndrome (CAPS) that completely resolved with plasmapheresis, intravenous immunoglobulin (IVIG), cyclophosphamide, rituximab, and anticoagulation indicating the potential for use of immunomodulating therapy for the treatment of osteonecrosis in the setting of secondary APS in patients with SLE.
Case report
A 20-year-old African American woman with a history of SLE, APS, hypertension, and chronic kidney disease (CKD) presents with severe sudden onset right knee pain with inability to bear weight on the right leg.
The patient was initially diagnosed with SLE at age 16 when she presented with lupus nephritis and arthralgias that were treated with cyclophosphamide and steroids. She was diagnosed with APS 10 years after her initial presentation, when she was found to have deep venous thrombosis, positive lupus anticoagulant, and positive anticardiolipin antibody. Anticoagulation was then initiated to treat her APS; however, she continued to have thrombotic events due to noncompliance with anticoagulation therapy.
Three weeks prior to this admission, our patient was treated with plasmapheresis, IVIG, rituximab, and cylcophosphamide for recurrent ischemic pancreatitis.
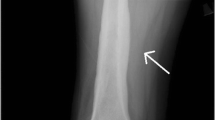
Evaluation of the right knee during this admission revealed multiple infarcts to the subchondral bone specifically in the posterior weight-bearing lateral femoral condyle, the subchondral bone of the medial femoral condyle at the junction of the weight-bearing and posterior flexion zone, the lateral femoral trochlear groove, in the superior aspect of the patella, and the largest in the proximal tibial diametaphysis—all confirmed by magnetic resonance imaging (MRI) (see Figs. 1, 2).

Fat-sensitive MRI (T1) and water-sensitive MRI (T2) with arrows indicating large areas of osteonecrosis in the femur

Fat-sensitive MRI (T1) and water-sensitive MRI (T2) with arrows indicating large areas osteonecrosis in the tibia and a small area in the distal femur indicated with a small arrow on T1
Her physical exam was unchanged from her baseline except for diffuse arthralgias and severe right knee pain with movement that limited her range of motion. The right knee was swollen, tender to palpation, with exacerbation of pain on flexion and extension. Left knee examination was normal. Muscle strength in all extremities remained normal.
Based on her clinical presentation, the patient was treated as a case of presumed CAPS given her history of recent ischemic pancreatitis, worsening renal failure, and now new onset of osteonecrosis. Therefore aggressive therapy with immunomodulating agents was initiated. Twelve sessions of plasmapheresis, IVIG, cyclophosphamide, and rituximab were given. IVIG was not given during previous admissions to prevent worsening of kidney function; however, with progression of the patient’s disease, it was decided at this time that the kidney function would be difficult to sustain and dialysis would be necessary in any case and so IVIG became a logical adjunct. With this therapy, the patient’s symptoms improved and eventually resolved.
After 16 months of follow up, all while the patient remained on anticoagulation and low dose prednisone (10–15 mg), she has not developed any further thrombotic events and continues to be ambulatory with ability to exercise without pain. Her kidney function deteriorated and she developed severe, refractory HTN with volume overload that required initiation of hemodialysis. Currently, the patient is doing well on dialysis, goes to college, and works 16 h per week. She is on a waiting list for a kidney transplant.
Discussion
Secondary APS in patients with SLE is a serious medical condition with high morbidity and mortality if not appropriately treated. Approximately 250,000 Americans have SLE and 20–35% of patients with SLE have APS [1, 2]. The disease is also known as antiphospholipid (aPL) syndrome, anticardiolipin antibody (aCL) syndrome, and lupus anticoagulant (LA) syndrome. APS is often found in the presence of other autoimmune diseases, such as SLE, in which case the disease is termed “secondary APS”. APS is defined as a pattern of recurrent thrombosis, pregnancy loss, and thrombocytopenia associated with the presence of a persistently positive aCL and/or LA test(s) [3]. The symptoms of APS include venous and arterial thromboses which can result in osteonecrosis; however, the thrombi more commonly cause deep vein thrombosis, pulmonary emboli, coronary syndrome, miscarriages, premature birth, and eclampsia. Neurological symptoms of APS include chronic headaches, stroke, vascular dementia, and seizures.
Current literature suggests that the cause of the clinical manifestations of APS stem mostly from the hypercoagulable state induced by the production of antibodies that bind to various phospholipids on components of the coagulation system. The resulting dysregulation of the coagulation system due to the pathogenic antibodies characteristic of APS, cause the thrombotic events that characterize APS.
The pathophysiology of osteonecrosis in patients with secondary APS has not been entirely established, however, many mechanisms have been proposed. These mechanisms revolve around two main ideas: first, anticardiolipin antibodies have been associated with thrombophilia and second, anticardiolipin antibodies have been associated with avascular necrosis of bone [4–13]. Therefore, osteonecrosis may be caused by thrombosis in the setting of a hypercoagulable state caused by the presence of antiphospholipid antibodies. We will first focus on the pathogenesis of the hypercoagulable state in the setting of APS, and then emphasize the pathogenesis of osteonecrosis in the hypercoagulable state generated by APS. We will then discuss the mechanism by which plasmapheresis and administration of IVIG and rituximab to our patient with secondary APS achieved complete resolution of the osteonecrosis.
A variety of mechanisms have been proposed to explain the hypercoagulable state found in patients with APS. One mechanism suggests that antiphospholipid antibodies inhibit activated protein C causing decreased inhibition of the clotting cascade [14–17]. Another mechanism suggests that antiphospholipid antibodies bind to platelets and promote platelet activation [18–20]. Furthermore, another proposed mechanism is that antiphospholipid antibodies interact with endothelial cells and induce expression of adhesion molecules and monocyte adhesion resulting in tissue factor expression and a procoagulant state [21–26].
Overall, the proposed mechanisms revolve around the idea that a hypercoagulable state predisposes a patient to vascular pathology resulting in bone death. While it is well known that thrombophilic and hypofibrinolytic coagulation abnormalities predispose patients to osteonecrosis, APS has been specifically implicated in generating a hypercoagulable state that predisposes the patient to osteonecrosis [27, 28]. The thrombophilic and hypofibrinolytic states generated by APS predispose patients to venous thrombi. The resulting venous occlusion of the bone by fibrin clots, leads to venous sinusoidal hypertension within the cancellous bone [29–31]. The venous sinusoidal hypertension increases to the point that arterial blood flow to the region is no longer able to deliver adequate oxygen to the bone. The resulting cellular hypoxia presumably gives way to bone and marrow cell death, thereby causing osteonecrosis [32]. This mechanism is consistent with the research finding that an elevated anticardiolipin antibody titer predisposes patients to osteonecrosis [33]. It has been shown that the combined use of daily plasmapheresis and rituximab is associated with clinical resolution of complications of thrombophilic disease states such as autoimmune thrombotic thrombocytopenic purpura, in which antibodies have been shown to be the offending agent [34]. The mechanism by which a combination of the administration of rituximab and plasmapheresis is able to bring about the resolution of the effects of an autoimmune disorder has been explored in other autoimmune diseases. Rituximab is a humanized monoclonal antibody raised against CD20 that leads to complement- and cell-mediated destruction of CD20-expressing cells. The B cells and pre-B cells express leukocyte antigens, CD19 and CD20, and then lose their expression of CD20 when they differentiate into antibody-secreting plasma cells. Although rituximab does not lead to removal of existing antibody-producing plasma cells, it eliminates the next generation of antibody-producing cells and prevents expansion of the reactive process [35]. Accordingly, the proposed mechanism of rituximab in the treatment of APS is believed to be the destruction of the CD20+ precursors of B cells that produce antiphospholipid antibodies resulting in decreased thrombophilia. IVIG inactivates currently circulating antibodies, and plasmapheresis removes antibodies from the blood and thereby reduces antibody titers, including anticardiolipin antibody, beta2-glycoprotein I antibody [36–38]. In this report, the patient experienced total recovery from knee osteonecrosis with administration of plasmapheresis, IVIG, cylcophosphamide, and rituximab.
The diagnosis of APS rests upon clinical evidence of vascular thrombosis and repeated positive tests for antibodies against phospholipids at least 6 weeks apart [1]. In patients who already have a diagnosis of APS with or without underlying SLE, reported symptoms of insidious joint pain should trigger a workup for osteonecrosis. Laboratory tests for osteonecrosis are extremely limited and their utilities are low. However, it is necessary to rule out more common etiologies that could potentially cause osteonecrosis such as sickle cell anemia and other hypercoagulopathies such as protein C and S deficiencies and factor V Leiden disease. Imaging modalities are preferred in the diagnosis of osteonecrosis. Serial plain radiographs can offer some insight into the progression of osteonecrosis, however, earliest signs of osteonecrosis can be detected with high specificity and sensitivity using MRI. On T1-weighted images, marrow fat of normal bone usually produces high signal intensity, which can be replaced by areas of low signal intensity in the early stages of osteonecrosis development, as seen in our figures [39].
Prophylactic prevention of thrombosis in patients with APS is warranted when patients have had previous episodes of vascular thrombosis. Warfarin is the agent of choice for anticoagulation and INR should be maintained between 3 and 4 to avoid further thrombotic complications. In patients with secondary APS, if there is a clinical suspicion of osteonecrosis, the utilization of MRI allows an early confirmation before the initiation of therapy. Treatments for osteonecrosis include both operative and nonoperative measures utilizing pharmacological approaches; only comparative pharmacotherapies will be discussed in this review.
There are several treatment options for CAPS, but no definitive regimen. As the number of patients with CAPS worldwide is small, no prospective, randomized, controlled trials on therapeutic strategies have been performed [40]. Suggested first-line therapies include anticoagulants and corticosteroids; plasmapheresis and IVIG are second-line treatments. Antihyperlipidemic agents and bisphosphonates have been used in the past for the treatment of symptomatic secondary osteonecrosis; the efficacy of these agents in improving the clinical outcome of osteonecrosis has been questionable [5]. Other treatments have been described in the literature, such as the use of rituximab, an anti-CD20 monoclonal antibody that acts against B cells; however, the use of these alternate modalities has been limited to a few small case series [41].
In this report, osteonecrosis in the patient’s right knee responded very well to the addition of immunomodulating therapies including plasmapheresis, IVIG, cylcophosphamide, and rituximab to anticoagulation with an excellent long-term clinical outcome. Knowing that the presence of antiphospholipid antibodies in APS is an independent risk factor for developing osteonecrosis, perhaps the ability of this regimen to remove these antibodies from the circulation and halt their production is contributing to the positive clinical outcome in treating this condition. Currently, rituximab is not indicated for the treatment of osteonecrosis; however, the significant recovery in our patient does encourage more trials of empiric therapy with rituximab for the treatment of osteonecrosis secondary APS in patients with and without SLE.
Conclusion
The treatment of osteonecrosis due to APS or CAPS in patients with SLE with immunomadulating therapy including plasmapheresis, IVIG, and rituximab can likely lead to resolution of symptoms and decrease morbidity and mortality in these patients. We arrive at this conclusion because this regimen decreases thrombophilia secondary to APS by way of two distinct mechanisms that share the common pathway of decreasing the thrombophilic antiphospholipid antibody titers. Further studies are needed to confirm this finding. However, the rapid deterioration of patients with this condition secondary to recurrent thrombosis can lead to very high morbidity and mortality rates. Therefore, clinical suspicion of the syndrome, aggressive response, and prompt imitation of therapy are mandated.
References
Abeles M, Urman JD, Rothfield N (1978) Aseptic necrosis of bone in systemic lupus erythematosus. Relationship to corticosteroid therapy. Arch Intern Med 138:750–754
Dimant J, Ginzler EM, Diamond HS et al (1978) Computer analysis of factors influencing the appearance of aseptic necrosis in patients with SLE. J Rheumatol 5:136–141
Miyakis S, Lockshin MD, Atsumi T et al (2006) International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 4:295–306
Gorshtein A, Levy Y (2007) Orthopedic involvement in antiphospholipid syndrome. Clin Rev Allergy Immunol 32:167–171
Agarwala S et al (2005) Efficacy of alendronate, a bisphosphonate, in the treatment of AVN of the hip. Rheumatology 44:352–359
Helmick CG, Felson DT, Lawrence RC, Gabriel S, Hirsch R, Kwoh CK et al (2008) Estimates of the prevalence of arthritis and other rheumatic conditions in the United States. Arthritis Rheum 58:15–25
Miyakis S, Lockshin MD, Atsumi I et al (2006) International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost 4:295–306
Bick RL, Baker WF (1992) Anticardiolipin antibodies and thrombosis. Hematol Oncol Clin North Am 6:1287–1299
Triplett DA (1993) Antiphospholipid antibodies and thrombosis: a consequence, coincidence, or cause? Arch Pathol Lab Med 117:78–88
Bick RL, Baker WF (1994) The antiphospholipid and thrombosis syndromes. Med Clin North Am 78:667–684
Asherson RA, Khamashta MA, Ordi-Ros J et al (1989) The “primary” antiphospholipid syndrome: major clinical and serological features. Medicine 68:366–374
Nagasawa K, Ishii Y, Mayumi T et al (1989) Avascular necrosis of bone in systemic lupus erythematosus: possible role of haemostatic abnormalities. Ann Rheum Dis 48:672–676
Alijotas J, Argemi M, Barquinero J (1990) Kienbock’s disease and antiphospholipid antibodies. Clin Exp Rheumatol 8:297–298
Seleznick MJ, Silveira LH, Espinoza LR (1991) Avascular necrosis associated with anticardiolipin antibodies. J Rheumatol 18:1416–1417
Vela P, Batlle E, Salas E, Marco P (1991) Primary antiphospholipid syndrome and osteonecrosis (letter). Clin Exp Rheumatol 9:545–546
Asherson RA, Liote F, Page B et al (1993) Avascular necrosis of bone and antiphospholipid antibodies in systemic lupus erythematosus. J Rheumatol 20:284–288
Migliaresi S, Picillo U, Ambrosone L, Di Plama G et al (1994) Avascular necrosis in patients with SLE: relation to corticosteroid therapy and anticardiolipin antibodies. Lupus 3:37–41
Oosting JD, Derksen RHWM, Bobbink IWG et al (1993) Antiphospholipid antibodies directed against a combination of phospholipids with prothrombin, protein C or protein S: an explanation for their pathogenic mechanism? Blood 83:2618–2625
Marciniak E, Romond EH (1989) Impaired catalytic function of activated protein C: a new in vitro manifestation of lupus anticoagulant. Blood 74:2426–2432
Malia RG, Kitchen S, Greaves M, Preston FE (1990) Inhibition of activated protein C and its cofactor protein S by antiphospholipid antibodies. Br J Haematol 76:101–107
Ieko M, Ichikawa K, Triplett DA (1999) β2-glycoprotein I is necessary to inhibit protein C activity by monoclonal anticardiolipin antibodies. Arthritis Rheum 42:167–174
Escolar G, Font J, Reverter JC (1992) Plasma from systemic lupus erythematosus patients with antiphospholipid antibodies promotes platelet aggregation. Arterioscler Thromb 12:196–200
Campbell AL, Pierangeli SS, Wellhausen S, Harris EN (1995) Comparison of the effects of anticardiolipin antibodies from patients with the antiphospholipid syndrome and with syphilis on platelet activation and aggregation. Thromb Haemost 73:529–534
Khamashta MA, Harris EN, Gharavi AE (1988) Immune mediated mechanism for thrombosis:antiphospholipid antibody binding to platelet membranes. Ann Rheum Dis 47:849–854
Kornberg A, Blank M, Kaufman S, Shoenfeld Y (1994) Induction of tissue factor-like activity in monocytes by anti-cardiolipin antibodies. J Immunol 153:1328–1332
Simantov R, LaSala JM, Lo SK (1995) Activation of cultured vascular endothelial cells by antiphospholipid antibodies. J Clin Invest 96:2211–2219
Del Papa N, Guidali L, Sala A (1997) Endothelial cells as target for antiphospholipid antibodies—human polyclonal and monoclonal anti-β2 glycoprotein I antibodies react in vitro with endothelial cells through adherent β2 glycoprotein I and induce endothelial activation. Arthritis Rheum 40:551–561
Cuadrado MJ, López-Pedrera C, Khamashta MA (1997) Thrombosis in primary antiphospholipid syndrome—a pivotal role for monocyte tissue factor expression. Arthritis Rheum 40:834–841
Amengual O, Atsumi T, Khamashta MA, Hughes GRV (1998) The role of the tissue factor pathway in the hypercoagulable state in patients with the antiphospholipid syndrome. Thromb Haemost 79:276–281
Vega-Ostertag M, Casper K, Swerlick R et al (2005) Involvement of p38 MAPK in the up-regulation of tissue factor on endothelial cells by antiphospholipid antibodies. Arthritis Rheum 52:1545–1554
Jones LC, Mont MA, Le TB et al (2003) Procoagulants and osteonecrosis. J Rheumatol 30:783–791
Mont MA, Ulrich SD, Seyler TM (2007) Role of thrombotic and fibrinolytic alterations in the pathogenesis and treatment of osteonecrosis. J Rheumatol 34:466–468
Glueck CJ, Freiberg R, Glueck HI et al (1994) Hypofibrinolysis: a common, major cause of osteonecrosis. Am J Hematol 45:156–166
Glueck CJ, Rorick MH, Schmerler M et al (1995) Hypofibrinolytic and atherogenic risk factors for stroke. J Lab Clin Med 125:319–325
Glueck CJ, Glueck HI, Greenfield D et al (1994) Protein C and S deficiency, thrombophilia, and hypofibrinolysis: pathophysiologic causes of Legg-Perthes disease. Pediatr Res 35:383–388
Gruppo R, Glueck CJ, McMahon RE, Bouquot J, Rabinovich BA, Becker A, Tracy T, Wang P (1996) The pathophysiology of alveolar osteonecrosis of the jaw: anticardiolipin antibodies, thrombophilia, and hypofibrinolysis. J Lab Clin Med 127:481–488
Mont MA, Glueck CJ, Pacheco IH, Wang P, Hungerford DS, Petri M (1997) Risk factors for osteonecrosis in systemic lupus erythematosus. J Rheumatol 24:654–662
Darabi K, Berg AH (2006) Rituximab can be combined with daily plasma exchange to achieve effective B-cell depletion and clinical improvement in acute autoimmune TTP. Am J Clin Path 125:592–597
Allford S, Machin S (2003) Guidelines on the diagnosis and management of the thrombotic microangiopathic haemolytic anaemias. Br J Haematol 120:556–573
Otsubo S, Nitta K, Yumura W, Nihei H, Mori N (2002) Antiphospholipid syndrome treated with prednisolone, cyclophosphamide and double-filtration plasmapheresis. Intern Med 41:725–729
Aaron RK (1998) Osteonecrosis: etiology, pathophysiology, and diagnosis. The adult hip, Lippincott-Raven Publishers, Philadelphia, pp 451–466
Acknowledgments
There is no conflict of interest in this document.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Haque, W., Kadikoy, H., Pacha, O. et al. Osteonecrosis secondary to antiphospholipid syndrome: a case report, review of the literature, and treatment strategy. Rheumatol Int 30, 719–723 (2010). https://doi.org/10.1007/s00296-009-1269-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00296-009-1269-9