
Abstract
The objective of this paper is to develop a computational model of the fission yeast (Schizosaccharomyces pombe) cell cycle using agent-based modeling (ABM), to study the sequence of states of the proteins and time of the cell cycle phases, under the action of proteins that regulate its cell cycle. The model relies only on the conceptual model of the yeast cell cycle regulatory network, where each protein has been represented as an agent with a property called activity that represents its biological function and a stochastic Brownian movement. The results indicate that the simulated phase time did have similar results in comparison with other models using mathematical approaches. Similarly, the correct sequence of states was achieved, and the model was run under different initial states to understand its emergent behaviors. The cell reached the G1 stationary state 94% of the times when running the model under biological initial conditions and 87% of the times when running the model through all the different combinations of initial states. Such results imply that the cell was capable to fix toward the biological expected phenomena. These results show that ABM is a suitable technique to study protein–protein interactions without using, often unavailable, kinetic parameters, or differential equations. This model sets as a base for further studies that involve the cell cycle of the fission yeast, with a special attention to studies and development of drug treatments for specific types of cancer.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The cell division cycle is a crucial process that occurs when the cell divides and reproduces. The cell cycle is a correct sequence of well-defined events. It starts with the duplication of the cell and its components and is followed by its division into two daughters with the same properties and capabilities as its mother. To complete the cell cycle, each cell has its own elaborate control and mechanisms that guarantee that the cell cycle is successful (Novak et al. 1998). In this paper, we use a computational approach to study the sequence of events and phase time duration of the fission yeast cell cycle without using kinetic parameters or differential equations, to develop a model that can be used as a base to further studies of the yeast cell cycle.
The sequence of events in eukaryotes is controlled by different proteins, whose interactions, concentrations, and molecular properties guide the cell to accomplish a correct full cell cycle (Csikász-Nagy et al. 2007). This cycle is divided into four phases: the Gap 1 (G1) phase, the Synthetic (S) phase, the Gap 2 (G2) phase, and the Mitotic (M) phase. In the G1 phase, the cell grows until reaching the necessary conditions to start the cell cycle, whereas in the S phase, deoxyribonucleic acid (DNA) is synthesized and the chromosomes are replicated. Then, in G2, the other components necessary to move to the M phase are synthesized. Next, in the M phase, the cell enters mitosis and divides itself. After the M phase, the cell enters a G1 stationary state, thus completing one cycle and waiting to have a division again (Davidich and Bornholdt 2008).
One of the most important model organisms for the study of eukaryotic cellular processes is the fission yeast (i.e., Schizosaccharomyces pombe). It is a relatively simple fungus and has a common eukaryotic cell cycle with nuclear mitotic divisions (Egel 2010). The fission yeast has been of great interest to scientists due to its cellular biology, sexual cycle, and the processes of growth and division that occur in its cell cycle (Egel 2010; Fantes and Hoffman 2016).
The study of the genetic control of cell division allowed to forge a very relevant fact within the cellular and molecular biology, the conservation of proteins and their functions through evolution. Where the genes that codify for the protein kinases, myosin or anthranilate synthases of newer organisms, are similar to the genes of those proteins in simpler organisms. Although the molecular conservation of the proteins involved in cell cycle mechanisms is not maintained between eukaryotes and prokaryotes, among the eukaryotes themselves, the similarity is incredibly significant. The main organisms studied in relation with the conservation of proteins within the cell cycle are Xenopus, S. pombe, and Saccharomyces cerevisiae, which have clear differences, but do resemble in that they code for the same dependent protein kinase on cyclins, one of the main molecules in the regulation of the cell cycle (Hartwell 2005).
One approach proposed by Hartwell (2005) to identify drugs with therapeutic advantage to treat specific types of cancer in humans, taking advantage of to the similarity in cell cycle process in the fission yeast and humans, is by constructing genetically modified yeast cells with mutations to match the behavior of the cancer cell. Then, the mutated yeast and a normal yeast used as a control are treated with different drugs to see which one killed the mutated cell faster, thus identifying a treatment that is specific for that type of cancer (Hartwell 2005).
The fission yeast cycle is often modeled from different perspectives and by considering multiple aspects. For instance, in their work, Novak et al. (1998) modeled the fission yeast cell cycle by considering cycle checkpoints, using differential equations, and protein kinetic parameters. Likewise, in Sveiczer et al. (2000), the authors developed a mathematical model and measured quantized cycle times with stochastic kinetic parameters. In addition, in Novak et al. (2001), the authors employed 14 differential and algebraic equations, whereas (Anbumathi et al. 2011) used differential equations to study the role of phosphatases in cell cycle regulation. Similarly, the budding yeast was modeled using stochastic Petri nets in Mura and Csikász-Nagy (2008), whereas researchers in Davidich and Bornholdt (2008) proposed a Boolean network model to predict the cell cycle sequence of the fission yeast.
Even though mathematical approaches are effective tools for studying fission yeast biological processes, computational biology techniques are becoming increasingly useful in the study and understanding of non-linear molecular mechanisms that gather complex protein-to-protein interaction networks (Wang 2016). Agent-based modeling (ABM) is the representation of autonomous agents or entities that are capable of making their own decisions. The decision-making process in ABM is defined by a set of rules for each agent. These rules are set according to the phenomenon that the agents represent. Then, the multiple interactions that occur between such agent rules enable to explore emergent phenomena and dynamics that cannot be studied using mathematical methods (Bonabeau 2002).
Agent-based modeling is a technique for studying biological systems. It can combine quantitative and qualitative information in the same model. Agent-based models are thus more complete, since they combine behavior rules (qualitative information) with numerical or mathematical data (quantitative information) about the phenomena to be explored (Bayrak et al. 2016). As a result, ABM is widely employed across disciplines (Bauer et al. 2009; Bayrak et al. 2016; Khataee et al. 2011; Manzanarez-Ozuna et al. 2015; Wang et al. 2015; Zhang et al. 2009). Furthermore, ABM assumes that everything can be modeled as long as there are agents, an environment, and interactions among agents and between agents and the environment (Wilensky and Rand 2015). Under these premises, ABM is capable of modeling cell cycle processes correctly if the appropriate conceptual model is employed, and as long as the agents and their interactions are properly defined.
This work develops a cell cycle agent-based model (CCABM) of the fission yeast and compares it in two ways. First, we compare our time results with those reported by Novak et al. (1998), as regards ideal phase times, and those reported in Novak et al. (2001) and Anbumathi et al. (2011), as regards phase time simulations. Then, the model behaviors under various circumstances are compared with the Boolean network model proposed in Davidich and Bornholdt (2008). Similarly, the CCABM was developed using neither kinetic parameters nor protein concentrations in the cell. This approach reduces the complexity of the conceptual model as well as the behavior and interaction rules of the proteins. Moreover, we relied on the qualitative representation of protein interactions (phosphorylation/dephosphorylation, binding, inactivation, activation, degradation) and ran the model under multiple initial states, as in Davidich and Bornholdt (2008).
Methodology
The creation process of the CCABM was divided into design, build, and analysis, based on Chapter 4 (pages 157–197) from Wilensky and Rand (2015).
Model design
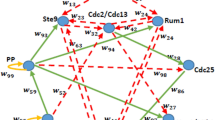
The conceptual model of the CCABM was designed according to the cell cycle regulatory network shown in Fig. 1 and proposed by Davidich and Bornholdt (2008). Davidich and Bornholdt’s proposal was constructed gathering information from Novak et al. (2001), Sveiczer et al. (2000), and Tyson et al. (2002). In that network, the nodes represent the regulatory proteins of a Boolean network, where each protein has two different states—0 (absent) or 1 (present)—that represent the capability of each protein to accomplish their biological function due to possible biological mechanisms, such as gene expression or protein phosphorylation. That network is capable to obtain the correct sequence of the cell cycle phases, but lacks in trying to measure phase times. To allow phases time calculation, the movement of the proteins was taken into account, where the proteins move with every time step, instead of only changing the states of the proteins according to the connection of the protein nodes.

Cell cycle regulatory network of the fission yeast
Cdc2/Cdc13 and Cdc2/Cdc13* are two different states of a cyclin-dependent protein kinase complex, being Cdc2/Cdc13 the primary protein involved in the control of the cell cycle. The remaining proteins are categorized into two groups. The positive regulators that include “Start” that works as a cell mass indicator: “Starter Kinases” (SK), which include proteins Cdc2/Cig1, Cdc2/Cig2, and Cdc2/Puc1; and the Cdc25 phosphatases. On the other hand, the antagonists of Cdc2/Cdc13 that are Slp1, Rum1, Ste9, a protein phosphatase (PP), and Wee1 and Mik1. However, Wee1 and Mik1 are grouped together as Wee1/Mik1, since they have the same function (Davidich and Bornholdt 2008). The initial states for the proteins were set as biological conditions that are: “Start” (1), SK (0), Cdc2/Cdc13 (0), Ste9 (1), Rum (1), Slp1 (0), Cdc2/Cdc13* (0), Wee1/Mik1 (1), Cdc25 (0), and PP (0). These initial states are the proteins’ states in the cell before the cell cycle starts. The cell has thus the necessary requirements to start and is waiting for the activation of SK by “Start”. Each protein or protein complex is represented by an agent in the model, where each of them is a different breed. Therefore, the model has a total of ten different agents (“Start”, SK, Cdc2/Cdc13, Ste9, Rum1, Slp1, Cdc2/Cdc13*, Wee1 Mik1, Cdc25, and PP) that coexist in the same environment: the inside of the cell.
Each agent has the property of being active (1) or inactive (0), which implies that it could be available for the cell cycle’s biological function that is accomplished when the biomolecules are in the same space (Lavalette et al. 2006), this is represented as Brownian dynamics, where the movement of two molecules is simulated as a succession of small stochastic displacements (Northrup and Erickson 1992), reasoning for why the agents have a stochastic type of movement in the environment. In turn, this means that the proteins’ or agent’s intercellular movement is based on a random function that chooses one out of eight possible orientations to move forward in every time step of the simulation. Agents can generate activation, inhibition, activation and inhibition, or self-degradation processes to other agents. An activation process will generate a change from 0 to 1 on another protein, whereas an inhibition process will generate a change from 1 to 0 on another protein. Finally, a self-degradation process will generate a change from 1 to 0 on the same protein.
The cell cycle regulatory network from Davidich and Bornholdt (2008) depicts all the interactions among proteins, molecules, or entities that regulate the cell cycle. In Fig. 1, the green arrows represent an activation process, the red arrows represent an inhibition process, and the yellow arrows represent an auto-inhibition process. In addition, notice that the model inputs are the states of each protein that is represented in the interface by binary selectors. These selectors define the initial inactivity (0) or activity (1) value of each protein (agent). As for the output, it will be the sequence of the cell cycle phases, the duration of each phase, and the final state of each protein.
Model construction
This step involved programming the CCABM according to the conceptual model defined in “Model design”. To this end, we relied on software NetLogo, version 6.1 (Wilensky 1999). First, a “setup” environment was created to define all the agents with their respective characteristics and properties. Interactions were programmed in the “go” environment, where these interactions are activation and inactivation processes that occur between two connected proteins (e.g., SK with Rum1, PP with Ste9). They generate state changes when both proteins react in the same physical space. To accomplish this, the “let” function was used, creating a local variable called “react” that represent the protein that is being affected (e.g., Rum1 when SK interacts with it). All the codes and NetLogo file of the model were uploaded to the CCABM’s GitHub repository https://github.com/biocastro/CCABM (Castro 2018).
Figure 2 shows the NetLogo interface of the model. The selectors for the initial conditions of the proteins are located on the top of the world. Left of the world, there is a selector to choose if links are visible or not. The setup button is to set the model with the initial states selected in the selector for initial conditions. The “bio” button is to set the initial states the same as the initial biological conditions ignoring the values set in the selector for initial states. The “go once” button is used to run one-time step and the go button is used to run the model entirely until the simulation is finished. At the right of the world are three rows of monitors, the first row shows the current phase, where the cell cycle is, the second row shows the time in ticks for each phase, and the third row shows the percentage of duration for each phase as well. Under the world are monitors that show the current value of activity for each protein. In the figure, the CCABM reached the G1 stationary state.

NetLogo interface of the cell cycle agent-based model
Analyzing the model
Models with stochastic properties should be run multiple times, to eliminate the inherent variability, to properly identify their behavior. If a model is run only once, it will be difficult to determine whether the results from that run indicate normal behavior or an atypical case. Therefore, to check phase durations, 100 run simulations were performed with the above-mentioned initial states. We measured the time at each run and calculated the average for all the runs. Then, we calculated the percentage of duration of each phase to compare our results with those reported by Sveiczer et al. (2000) and Novak et al. (2001), main references for the creation of the cell cycle regulatory network performed by Davidich and Bornholdt (2008), and with the findings of Novak and Tyson (1995) and Anbumathi et al. (2011).
To understand the behavior of the CCABM, three sets of experiments similar to Davidich and Bornholdt (2008) were run. The experiments were set and run using the Behavior Space tool from NetLogo, which allows a specific experiment to be set by defining input values variation to run the model under the same conditions as many times as needed. For the first experiment, initial states were set as mentioned above. To discard anomalous behavior, 100 runs were performed when checking the states of the proteins during the whole cell cycle simulations until reaching the G1 phase. In this phase, Ste9, Rum1, and Wee1/Mik1 have an activity value of 1, whereas the other proteins have an activity value of 0. When reaching the G1 phase, the cell is waiting for another cycle to start.
The second experiment was performed by running the model from the 210 = 1024 combinations or possible initial states of the proteins. The goal was to verify whether the different fixed points were reached (where the model stops) and to calculate the percentage of cases, where the G1 stationary state was attained. This allowed us to determine whether the cell was fixing toward the expected biological outcome. Finally, 512 more runs were performed, starting from those initial conditions, where “Start” was active again. The purpose was to calculate the percentage of cases, where the cycle ended in the G1 stationary state. The goal of this third set of experiments was to analyze how the model behaves when the conditions are appropriate to start the cycle and the proteins are not in the initial expected conditions.
Results
The results of the first 100 runs are reported in Table 1 for each phase. The table also includes the results reported in the literature to provide a comparison. The runs were performed to measure phase time in an average of tick’s duration (computational time to finish one code iteration and the way NetLogo measures time progression). Minor differences between models are expected, nevertheless, all of them have a specific outcome, the G2 dominance over the other three phases, characteristic behavior of the fission yeast cell cycle as mentioned in Novak et al. (1998).
To make a direct comparison, we calculated the percentage of duration over the total time of the cell cycle for each phase. Figure 3 depicts the time duration percentage for the five models. When comparing the CCABM with the findings reported by Anbumathi et al. (2011), we found no significant difference in G1, S, and M phases. Similar to the results reported by Sveiczer et al. (2000) having no significant difference in G1 and G2 phases. Despite not having the exact same behavior between all phases, the CCABM showed similar behavior to that two models were G2 > G1 > M > S according to their prediction of phase time. On the other hand, when comparing the CCABM to the models proposed by Novak et al. (2001) and Novak and Tyson (1995), the difference in phase time was bigger, and also having a different overall behavior, where G2 > G1 > S > M phase time-related. Nevertheless, all models have a good approximation to the biological phenomenon of the fission yeast cell cycle.

Phase time duration comparison among fission yeast cell cycle models
Table 2 reports the main course of events leading to a correct and normal cell cycle. As can be observed, the last state belongs to the fixed point of the G1 stationary state, which is the most important state for the cell to reach. In addition, the CCABM reached the G1 stationary state in 94% of the cases. On the other hand, Table 2 reports the sequence of events throughout the four phases, starting in the G1 phase and finishing in the M phase, to enter the G1 stationary state again. Notice that this outcome is based on the network’s connection and its conversion to the computational model, thereby rejecting any type of linearity due to model stochasticity.
To understand the different possible results and fixed points of the regulatory proteins at the end of the cycle, the model was run over 1024 possible initial states. Table 3 shows all the different states obtained after running the above-mentioned experiments and the number of times it happened out of the 1024 runs. In this case, the G1 stationary state was reached 87% of the times. Similar results are reported in Davidich and Bornholdt (2008). Therefore, our findings indicate that the cell cycle will often move toward the G1 stationary state in the presence of perturbations affecting proteins, as it occurs in the biological cell cycle of the fission yeast.
Table 3 shows a fixed point, where all the proteins are inactive. Overall, it is the same state, where Cdc25 and PP are active. As the regulatory networks describe, PP inactivates Cdc25, but it also inactivates itself. Therefore, the two proteins move from an active state to an inactive state really close in the sequence. This is caused by the stochastic nature of the ABM and does not represent a failure in the model. Finally, when running the model in the different initial states, where “Start” was active, 91% of the times the cycle reached the desired G1 stationary phase. This is reliable evidence that the cell cycle will follow the correct sequence of events when the cell is ready to start the cycle. The fact that the model did not reach the desired state 100% of the times implies that the model has stochastic properties, thus resulting in a model that works with both network design and randomness, and not only through a strict set of rules that define the sequence of events.
Discussions
The results of the comparative analysis between the CCABM of the fission yeast and other phase time simulation models were highly promising. All the models report similar behaviors and phase duration period percentages with respect to the theoretical characteristic behavior of the fission yeast cell cycle. Such findings imply that the CCABM correctly simulates the phase times of the biological yeast cell cycle without relying on differential equations, kinetic parameters, or concentrations from proteins in the regulatory network of the cell cycle.
A comparison between two modeling techniques was performed to determine the feasibility of ABM in the study of the fission yeast’s cell cycle. Our results were similar to those reported by Davidich and Bornholdt (2008), whose model had a behavior similar to the biological phenomena, even when tested under different initial conditions. This means that the network interaction and programming are correct due to the flow of states reaching the G1 stationary state. It is important to notice the main advantage of the CCABM against the Boolean network model, where the latter lacks the ability to predict the time duration for each phase, contrary to the CCABM that can fully reproduce the sequence of states during the cell cycle and measure the duration of each phase.
As previously mentioned, the CCABM was developed without considering kinetic parameters for the molecular processes involved in the fission yeast cell cycle. This represents an advantage of ABM if compared to other mathematical models. Kinetic parameters are often not available in the literature, and their measurement tends to be time-consuming and resource consuming. Another advantage of the CCABM is its ability to measure both phase times and sequence for the cell cycle. Therefore, ABM seems to be a viable approach to the study of the fission yeast’s cell cycle. Likewise, ABM might be able to model any other type of behavior that is represented by a network of molecules interacting with others within the same network (e.g., protein–protein interactions). In addition, both ABM and Boolean networks have an advantage over differential equations, as they focus only on the qualitative interaction of the proteins.
This research recommends the use of ABM, since it has a wide range of applications across disciplines. Moreover, researchers do not need to be software experts, and ABM can be easily performed thanks to the Behavior Space tool that allows performing a number of experiments to be run in different conditions. In addition, the CCABM can effectively support the study of the cellular process in the fission yeast, which is a useful biological model for cell cycle regulation.
The CCABM aims to be a base to further models that involve fission yeast cell cycle studies. One of its possible applications is in the development of specific treatments for different types of cancer. That can be achieved by changing the behavior of the regulatory network to match the behavior of a specific type of tumor, and then adding an agent or set of agents that represent the anticancer treatment to see if that makes the yeast cell to die, thus, reducing time and materials used in the laboratory. The properties of ABM and NetLogo give scientist a breach to easily modify the CCABM to performed the changes mentioned above, being the only impediment the knowledge about the type of cancer to be studied and the properties of the drugs to be tested. Nevertheless, with the appropriate information of the fission yeast cell cycle, the behavior of the type of cancer and the molecular mechanisms involved in the success of the anticancer drug to be applied to the CCABM could create a successful model for that case study.
Conclusions
This work proposes an agent-based model for the fission yeast cell cycle. As its major contribution, the model is able to reproduce the sequence of events in the cell cycle and the duration of each cycle phase (i.e., G1, S, G2, and M) without taking into account differential equations or kinetic parameters. Therefore, the model paves the way for new resource-saving and time-saving approaches to the study of the cell cycle. Similarly, this research demonstrates that systems biology is an effective approach to analyze biological phenomena that are difficult or impossible to study in laboratories. Hence, in the context of cellular and biochemical problems, systems biology, alongside ABM, is a promising field, not to replace in vivo experimentation but to enhance it and improve it with the help of in silico experiments.
The success of this model gives scientists a tool to develop other models to help in research involving fission yeast cell cycle studies.
References
Anbumathi P, Bhartiya S, Venkatesh KV (2011) Mathematical modeling of fission yeast Schizosaccharomyces pombe cell cycle: exploring the role of multiple phosphatases. Syst Synth Biol 5(3–4):115–129. https://doi.org/10.1007/s11693-011-9090-7
Bauer AL, Beauchemin CAA, Perelson AS (2009) Agent-based modeling of host-pathogen systems: The successes and challenges. Inf Sci 179(10):1379–1389. https://doi.org/10.1016/j.ins.2008.11.012
Bayrak ES, Wang T, Jerums M, Coufal M, Goudar C, Cinar A, Undey C (2016) In silico cell cycle predictor for mammalian cell culture bioreactor using agent-based modeling approach. IFAC PapersOnLine 49(7):200–205. https://doi.org/10.1016/j.ifacol.2016.07.249
Bonabeau E (2002) Agent-based modeling: methods and techniques for simulating human systems. Proc Natl Acad Sci 99(suppl. 3):7280–7287. https://doi.org/10.1073/pnas.082080899
Castro C (2018) biocastro/CCABM: cell cycle agent-based model of the fission yeast. https://doi.org/10.5281/ZENODO.1253360
Csikász-Nagy A, Kapuy O, Giorffy B, Tyson JJ, Novák B (2007) Modeling the septation initiation network (SIN) in fission yeast cells. Curr Genet. https://doi.org/10.1007/s00294-007-0123-4
Davidich MI, Bornholdt S (2008) Boolean network model predicts cell cycle sequence of fission yeast. PLoS One. https://doi.org/10.1371/journal.pone.0001672
Egel R (2010) The Molecular biology of Schizosaccharomyces pombe. Springer, Berlin. https://doi.org/10.1007/978-3-662-10360-9
Fantes PA, Hoffman CS (2016) A brief history of Schizosaccharomyces pombe research: a perspective over the past 70 years. Genetics 203(2):621–629. https://doi.org/10.1534/genetics.116.189407
Hartwell LH (2005) Yeast and cancer. Biosci Rep 24(4–5):525–544. https://doi.org/10.1007/s10540-005-2743-6
Khataee HR, Aris TNM, Sulaiman MN (2011) An agent-based model of muscle contraction process as a bio-robotic process. In: 2011 5th Malaysian conference in software engineering, MySEC 2011, pp 55–60. https://doi.org/10.1109/MySEC.2011.6140643
Lavalette D, Hink MA, Tourbez M, Tétreau C, Visser AJ (2006) Proteins as micro viscosimeters: Brownian motion revisited. Eur Biophys J 35(6):517–522. https://doi.org/10.1007/s00249-006-0060-z
Manzanarez-Ozuna E, Flores DL, Gómez-Gutiérrez CM, Abaroa A, Castro C, Castañeda-Martínez RA (2015) Modelo basado en agentes de la vía MAPK con NetLogo. In: Memorias Del Congreso Nacional de Ingeniería Biomédica, (October), 5–10. https://doi.org/10.13140/RG.2.1.4179.6244
Mura I, Csikász-Nagy A (2008) Stochastic Petri Net extension of a yeast cell cycle model. J Theor Biol 254(4):850–860. https://doi.org/10.1016/j.jtbi.2008.07.019
Northrup SH, Erickson HP (1992) Kinetics of protein-protein association explained by Brownian dynamics computer simulation. Proc Natl Acad Sci 89(8):3338–3342. https://doi.org/10.1073/pnas.89.8.3338
Novak B, Tyson JJ (1995) Quantitative analysis of a molecular model of mitotic control in fission yeast. J Theor Biol 173(3):283–305. https://doi.org/10.1006/jtbi.1995.0063
Novak B, Csikasz-Nagy A, Gyorffy B, Chen K, Tyson JJ (1998) Mathematical model of the fission yeast cell cycle with checkpoint controls at the G1/S, G2/M and metaphase/anaphase transitions. Biophys Chem 72:185–200
Novak B, Pataki Z, Ciliberto A, Tyson JJ (2001) Mathematical model of the cell division cycle of fission yeast. Chaos 11(1):277–286. https://doi.org/10.1063/1.1345725
Sveiczer A, Csikasz-Nagy A, Gyorffy B, Tyson JJ, Novak B (2000) Modeling the fission yeast cell cycle: quantized cycle times in wee1− cdc25Delta mutant cells. Proc Natl Acad Sci USA 97(14):7865–7870. https://doi.org/10.1073/pnas.97.14.7865
Tyson JJ, Csikasz-Nagy A, Novak B (2002) The dynamics of cell cycle regulation. BioEssays 24(12):1095–1109. https://doi.org/10.1002/bies.10191
Wang Z (2016) Big data mining powers fungal research: recent advances in fission yeast systems biology approaches. Curr Genet. https://doi.org/10.1007/s00294-016-0657-4
Wang Z, Butner JD, Kerketta R, Cristini V, Deisboeck TS (2015) Simulating cancer growth with multiscale agent-based modeling. Semin Cancer Biol 30:70–78. https://doi.org/10.1016/j.semcancer.2014.04.001
Wilensky U (1999) NetLogo. Center for Connected Learning and Computer-Based Modeling, Northwestern University, Evanston, IL. http://ccl.northwestern.edu/netlogo/. Accessed Jun 2017
Wilensky U, Rand W (2015) An introduction to agent-based modeling. The MIT Press, Cambridge. https://mitpress.mit.edu/books/introduction-agent-based-modeling. Accessed Jan 2017
Zhang L, Wang Z, Sagotsky JA, Deisboeck TS (2009) Multiscale agent-based cancer modeling. J Math Biol 58(4–5):545–559. https://doi.org/10.1007/s00285-008-0211-1
Acknowledgements
Funding was provided by Consejo Nacional de Ciencia y Tecnología (Grant no. 763840).
Funding
Funding was provided by Consejo Nacional de Ciencia y Tecnología (Grant no. 763840).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there are no conflicts of interest with respect to the publication of this research.
Additional information
Communicated by M. Kupiec.
Rights and permissions
About this article

Cite this article
Castro, C., Flores, DL., Cervantes-Vásquez, D. et al. An agent-based model of the fission yeast cell cycle. Curr Genet 65, 193–200 (2019). https://doi.org/10.1007/s00294-018-0859-z
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-018-0859-z