
Abstract
Agrobacterium-mediated gene transfer (AMT) is extensively employed as a tool in fungal functional genomics and accordingly, in previous studies we used AMT on a dikaryotic strain of the ectomycorrhizal basidiomycete Laccaria bicolor. The interest in this fungus derives from its capacity to establish a symbiosis with tree roots, thereby playing a major role in nutrient cycling of forest ecosystems. The ectomycorrhizal symbiosis is a highly complex interaction involving many genes from both partners. To advance in the functional characterization of fungal genes, AMT was used on a monokaryotic L. bicolor. A collection of over 1200 transgenic strains was produced, of which 200 randomly selected strains were analyzed for their genomic T-DNA insertion patterns. By means of insertional mutagenesis, a number of transgenic strains were obtained displaying differential growth features. Moreover, mating with a compatible strain resulted in dikaryons that retained altered phenotypic features of the transgenic monokaryon. The analysis of the T-DNA integration pattern revealed mostly similar results to those reported in earlier studies, confirming the usefulness of AMT on different genetic backgrounds of L. bicolor. Taken together, our studies display the great versatility and potentiality of AMT as a tool for the genetic characterization of L. bicolor.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ectomycorrhiza (ECM) has a central role in the nutrient flow in temperate and boreal forest ecosystems. Numerous studies have demonstrated that while engaged in ECM interactions both the fungus and the host plant benefit from the nutrient and water exchange that occur at the symbiotic structures (Smith and Read 2008). The plant receives mainly nitrogen and phosphorus, while the fungus (predominantly basidiomycetes) is provided with carbon in the form of photosynthesis-derived metabolites. Also, it has been proven that the association of the plant with the fungus, whose hyphae spread over much larger extensions of soil than the tree roots alone, gives the former a better access to water as well as to macro- and micro-nutrients (Peterson et al. 2004; Lucic et al. 2008; Nygren et al. 2008; Tatry et al. 2009). Moreover, the interspecific and bidirectional belowground carbon trade between trees through ECM has recently been demonstrated and measured (Klein et al. 2016). This indicates that the forest trees interact with each other via symbiotic fungi in an even more complex manner than previously believed and a comprehensive understanding of the fungal mechanisms involved in the establishment and function of the ECM symbiosis is thus of great importance.
Studies of organisms and their relations with the environment are made through functional analyses, which allow the integration of different macro- and micro-scopic, physiological, biochemical, ecological and molecular features.
The development of ECM structures requires a fine space–time coordination in the gene expression of both the fungal and the plant partner (Wright et al. 2005). Also, the formation in the plant’s radical apoplast of a Hartig net resembles in certain points the invasion produced by pathogenic fungi. However, a distinctive characteristic of this mutualistic colonization that has long been observed and more recently proven is the absence of a pronounced defensive response from the plant (Martin et al. 2008). This implies that symbiotic fungi have specialized in suppressing and evading the plant’s innate defenses, through the production of secreted signal molecules such as effector proteins which can suppress the host immune response, or manipulate host cell physiology (Plett et al. 2011, 2014a, b; Lo Presti et al. 2015). The study of these complex mechanisms demands the existence of precise molecular tools that must be applicable to a wide range of experimental systems.
Gene transfer is a fundamental step in the genetic manipulation of fungi and it is an essential tool for functional genomic approaches. Several methods have been used for the introduction of foreign genetic material into fungal cells. This is a fundamental step in the genetic manipulation of these organisms, essential for functional genomic approaches. The more widely known techniques of electroporation and PEG-mediated transformation have been applied to different mushrooms (Li et al. 2006; Kuo and Huang 2008; Kim et al. 2010; Yin et al. 2012). Their efficiency greatly varies from fungus to fungus but they share a basic obstacle: the need to work with protoplasts. Forming viable protoplast from filamentous fungal material is a laborious process. Protoplasts are very delicate and need carefully monitored osmotic conditions to minimize cell mortality, and the post-transformation regeneration step of mycelia under selective conditions often brings further technical complications. The delivery of the foreign DNA into the fungal protoplasts is usually carried out in the form of non-autoreplicative molecules and the maintenance of this introduced material is dependent on genomic integration of the transgenes. Some autoreplicative vectors are available for gene transfer in fungi, especially for Aspergillus spp. (Aleksenko and Clutterbuck 1997; Carvalho et al. 2010), but the scarcity of naturally occurring plasmids strongly suggests that these are not very stable genetic elements in fungi.
The transformation mediated by Agrobacterium tumefaciens (AMT) offers an alternative method for genetic manipulation of fungi. AMT takes advantage of the natural capacity of this bacterium to transfer DNA to other organisms, in nature to a plant host, and stably integrate it into its genome. Agrobacterium carries a virulence plasmid which encodes both the mobile DNA element (referred as “T-DNA” in its double-stranded form and as “T-strand” in its single-stranded DNA form present during the mobilization), and the protein machinery needed for the transfer process. The T-DNA is flanked by 25 bp long direct, imperfect repeats called the right border (RB) and the left border (LB), and the key for using Agrobacterium as a gene delivery vehicle was the finding that the native T-DNA sequence between these borders could be modified without affecting the delivery or the integration process (Bourras et al. 2015).
Ectomycorrhiza is formed by filamentous basidio- and asco-mycetes, and the lack of efficient, protoplast-free, gene transfer method applicable to these fungi has seriously hindered the genetic studies of the ECM interaction.
While widely used in plant genetics since the early 1980s, Agrobacterium has been shown, under laboratory conditions, to be able to transfer its T-DNA also to non-plant cells, these including both yeasts and filamentous fungal species (Piers et al. 1996; Shi et al. 2012; Zhang et al. 2014; Aragona and Valente 2015). Furthermore, several ECM basidiomycete species have been demonstrated to be susceptible to AMT, opening totally new possibilities for ECM research (Pardo et al. 2002; Hanif et al. 2002; Müller et al. 2006; Zubieta et al. 2014). One of these fungi is Laccaria bicolor, the first ECM basidiomycete with its whole genome sequenced (Kemppainen et al. 2005; Martin et al. 2008).
We have previously examined the frequency and sequence bias of the T-DNA integration in the genome of the dikaryotic L. bicolor strain S238 N (Kemppainen et al. 2008). Similarly to plants, the T-DNA integration has been reported to occur preferably as a single copy per genome in fungi (Bundock et al. 1995; Alonso et al. 2003; Schneeberger et al. 2005; Zhang et al. 2007; Choi et al. 2007; Bourras et al. 2012). Our results from Laccaria AMT support the simple T-DNA integration pattern and revealed a bias towards coding sequence integrations in the fungal genome. However, no conserved nucleotide motifs were detected between the genomic sites of integration. All these characteristics confirmed AMT as a very appropriate tool for functional genomics in the dikaryotic strain. We have since demonstrated the usefulness of AMT for other objectives, such as gene expression downregulation and overexpression (Kemppainen et al. 2009; Kemppainen and Pardo 2010; Plett et al. 2011; Kemppainen and Pardo 2013; Navarro-Ródenas et al. 2015; Xu et al. 2015, 2016). In all instances, modification of a single nucleus was sufficient to achieve the desired goal. However, there are situations when it would be most useful to be able to obtain dikaryons that have both nuclei modified. Therefore, in the present work we analyzed the T-DNA integration pattern in an insertional mutagenesis library generated by AMT in the monokaryotic L. bicolor strain S238 N-H82. We obtained a collection of over 1200 transgenic strains, of which 200 randomly selected strains were subjected to transgene integration pattern analysis.
Materials and methods
Fungal and bacterial strains
Laccaria bicolor (Maire) Orton (Di Battista et al. 1996) monokaryotic strains S238 N-H82 and S238 N-H107 (Selosse et al. 1996) were used in this study. The monokaryotic strains were kindly provided by Dr François Le Tacon from INRA Nancy, France. The vegetative mycelia were maintained at 22 °C in the dark on Pachlewski (P5)-modified agar medium, according to Kemppainen et al. (2008). The transgenic S238 N-H82 strains, containing the T-DNA of the pHg/pBks binary vector, were cultured in the same medium, with the addition of 300 μg/ml hygromycin B (Invitrogen). Escherichia coli TOP10 (Invitrogen) was used for plasmid cloning. Agrobacterium tumefaciens AGL-1 was employed in Laccaria AMT.
AMT of L. bicolor
Laccaria bicolor S238 N-H82 was transformed with A. tumefaciens AGL-1 containing the pHg/pBks vector according to a previously established protocol (Kemppainen et al. 2005). The hygromycin resistance (hph) carrying transgenic fungal strains were passed three times on GPY medium (Mg2SO4·7H2O 0.5 g/l; KH2PO4 1 g/l; glucose 20 g/l; maltose 5 g/l; tryptone 5 g/l; peptone 5 g/l; yeast extract 3 g/l; agar–agar 20 g/l; pH: 5.5) with 300 μg/ml hygromycin B before growing mycelia for subsequent use.
Southern blotting
Fungal strains were grown on cellophane membranes on P5 medium with 300 μg/ml hygromycin B. The gDNA was purified with the DNeasy® Plant Mini Kit (Qiagen) and eluted with ddH2O. Approximately, 6 μg of fungal genomic DNA (gDNA) per sample was digested with SacI (Promega) at 37 °C overnight, separated in 1 % agarose gel and transferred by alkaline capillarity blotting to an Amersham™ Hybond™ N Blotting Membrane (GE Healthcare) according to the manufacturer’s protocol. A 1020 bp hph probe was generated from pHg/pBks by PCR using primers HpH-For/HpH-Rev (5′ AAGCCTGAACTCACC GCGAC 3′/5′ CTATTCCTTTGCCCTCGGAC 3′) (Invitrogen). The amplification was made according to Kemppainen et al. (2008) and the amplification product was gel purified using the QIAquick® Gel Extraction kit (Qiagen). The labeling of the probe, hybridization and signal detection were performed with the Gene Images AlkPhos Direct Labelling and Detection System (GE Healthcare), according to the manufacturer’s instructions, hybridizing at 55 °C overnight and detecting after 6 h of exposure to a film (Agfa New Medical X-ray film).
Vegetative growth screening
The hygromycin-resistant strains obtained by AMT were cultured on solid P5 and GPY media for comparison of growth rates with the wild type strain S238 N-H82. Each screened strain was grown as triplicates on 100 mm diameter Petri dishes with 20 ml of medium at 22 °C in darkness. The vegetative growth was determined by measuring the colony radius every 5 days for 30 days, taking three measures for each colony. Strains with growth rates different from the wild type on one or both media were selected for further studies.
For liquid culture studies, three 1 × 1 mm plugs of mycelium were used to inoculate 125 ml Erlenmeyer flasks containing 20 ml of P5 or GPY media and kept static at 22 °C in darkness. Dry weight was determined every 5 or 7 days for 35 days. For each strain, two independent studies were made, with three replicas for every determination.
Dikaryon formation
The monokaryotic transgenic strains selected based on the growth screening and the wild type strain were mated with the compatible strain S238 N-H107 wt by co-cultivation on solid P5 for 30 days at 22 °C in darkness. The mycelia from the areas of interaction were propagated on the same medium with the addition of 300 μg/ml hygromycin B. The formation of the dikaryon was confirmed by the observation of clamp connections under a Nikon Eclipse 200 microscope.
Plasmid rescue
Fungal strains were grown on cellophane membranes on P5 medium with 300 μg/ml hygromycin B. The gDNA was purified with the DNeasy® Plant Mini Kit (Qiagen) and eluted with ddH2O. To ascertain the integrity of the hygromycin and ampicillin resistance genes, each gDNA sample was subjected to two PCRs with primers HpH-For/HpH-Rev and Amp-For/Amp-Rev (5′ CCCAAGGTTTGCAAGCAGCAGATTACGCG 3′/5′ CGCGGATCCGCTCATGAGACAATAACCC 3′) (Invitrogen). The PCRs were carried out according to Kemppainen et al. (2008). One to 3 μg of each gDNA was digested with SacI (Promega) (20–30 U/μg DNA) overnight. The enzymatic reactions were heat-inactivated, precipitated with ammonium acetate/ethanol and dissolved in ddH2O (Sambrook et al. 1989). Cut gDNA was self-ligated with 1 U of T4 DNA ligase (Promega) in 50 μl final volume at 4 °C overnight and precipitated with ammonium acetate/ethanol. Samples were dissolved in 10 μl of ddH2O and 5 μl was used for electroporating E. coli by a standard protocol (Sambrook et al. 1989). Electroporated bacteria were plated onto LB agar medium supplemented with 100 μg/ml ampicillin. Six ampicillin-resistant bacterial colonies were picked up for each fungal strain and cultured in LB liquid medium with ampicillin overnight for plasmid purification. The plasmids were linearized with SacI (Promega), which cuts once within the rescued plasmids from the transgenic L. bicolor strains. Restriction products were separated in 1 % agarose gels and stained with ethidium bromide. For every fungal transformant, the size of the 6 plasmids was analyzed to identify possible multiple insertional events. If all plasmids had the same size, the analysis was continued with only one bacterial clone. For the fungal strains that failed to produce resistant bacterial colonies at first, the rescue was attempted again by increasing the amount of gDNA.
DNA sequencing and analysis of the T-DNA integration sites
The rescued plasmids were purified for sequencing with the QIAprep® Spin Miniprep Kit (Qiagen). The sequencing was performed with the Post-RB primer (5′-AGCAGCTTGAGCTTGGATC-3′) (Invitrogen), in a 3730XL DNA sequencer (Macrogen Inc, Seoul, Korea). The sequences obtained were compared with the L. bicolor genome using the BLASTN algorithm on the JGI genome portal (http://genome.jgi-psf.org/Lacbi2/Lacbi2.home.html). Upstream regions were defined as up to 1500 bp upstream from the start codon and downstream regions up to 500 bp downstream from the stop codon. Sequence alignments were performed using the CLUSTALW2 algorithm (http://www.ebi.ac.uk/Tools/msa/clustalw2/). The alignments were used to create a Sequence Logo with the WebLogo online software available at http://weblogo.berkeley.edu/logo.cgi.
Results
In a previous study, we demonstrated that AMT can be used for gene transfer in dikaryotic L. bicolor. The T-DNA integrates primarily as a single copy without sequence bias, making the method suitable for random mutagenesis of the entire fungal genome (Kemppainen et al. 2005, 2008). In the present study, we have used the plasmid rescue binary vector pHg/pBks to investigate the integration pattern in a monokaryotic genetic background of L. bicolor and extended the study to a larger transgenic library.
Laccaria bicolor monokaryon S238 N-H82 was transformed with Agrobacterium tumefaciens AGL-1 carrying the pHg/pBks vector. This vector has no sequence homology with the fungal genome and allows the selection of transgenic strains with hygromycin B. It contains the elements necessary for an easy identification of the integration site by plasmid rescue (Fig. 1). A collection of 1250 transgenic strains was obtained by AMT according to Kemppainen et al. (2005) using GPY medium (glucose-yeast extract-peptone) instead of P5. This change was made to allow the growth of insertional mutants with defective metabolic pathways.

T-DNA in pHg/pBk. LB T-DNA Left Border from pCAMBIA1300. HRC Hygromycin resistance cassette, composed by the glyceraldehyde 3-phosphate dehydrogenase promoter from Agaricus bisporus, the hygromycin B O-phosphotransferase (hph) coding sequence from E. coli (confers resistance to hygromycin B) and the 35S terminator from the cauliflower mosaic virus. SacI position of the SacI restriction enzyme recognition site. AmpR bla gene form E. coli, β-lactamase that confers ampicillin resistance. ori replication origin from pBlueScript KS + (pBks). POST-RB primer binding site. RB T-DNA right border from pCAMBIA1300
To ascertain if the tendency of single integrations found for the dikaryotic strain existed also in the monokaryon, a Southern blot assay was performed with 9 randomly selected hygromycin-resistant strains. After purification of the gDNAs, the presence of hph was tested by PCR, with positive results for every strain (data not shown). The genomes were cut using the restriction enzyme SacI, which has one recognition site inside the T-DNA sequence (Fig. 1), and hybridized with a PCR-generated hph probe. At least two-thirds of the strains had a single copy of the T-DNA integrated into the genome (Fig. 2). This percentage appears to be lower than in the case of the dikaryon [90 % single integration events, (Kemppainen et al. 2008)], but it is in keeping with the tendency of single T-DNA integrations.

Southern blot analysis of L. bicolor S238 N-H82 transgenic and wild type strains. From left to right, M molecular size marker (λ-EcoRI/HindIII); WT wild type strain; 1–9 Randomly selected transgenic strains from the library
The next step in the characterization of the integration pattern was the random selection of 200 out of the 1250 transgenic strains. Their gDNA was purified and subjected to PCR with primers specific for the ampicillin resistance cassette, present in the T-DNA. The 190 ampicillin-positive strains were further used for the plasmid rescue protocol according to Kemppainen et al. (2008).
Out of 190 ampicillin-positive fungal strains, 109 produced ampicillin-resistant bacterial colonies. Purified plasmids of the minimum size were then sequenced using the Post-RB primer (Fig. 1) yielding 99 successful reactions. The majority of these (87/99) contained identifiable sequences from the L. bicolor genome and had a significant degree of conservation of the RB (Fig. 3). The remaining 12 sequences corresponded to empty plasmidic vectors or did not carry enough genomic sequence to allow the identification of a unique integration site. Few sequences belonged to transposable elements, which are found throughout the fungal genome and could, therefore, not be mapped to a single locus. A BLASTN search was performed by means of the JGI Laccaria genome portal (http://genome.jgi.doe.gov/Lacbi2/Lacbi2.home.html) to identify the precise T-DNA–gDNA junctions. The integrations were mapped in intergenic or coding regions of the gene models annotated in the genome by automated predictions or manually curated.

Frequency plot of the right border of the T-DNA right border (RB)—gDNA junction in the L. bicolor S238 N-H82 transgenic strains. All the nucleotides that appear in a given position are shown. The height of each letter represents the frequency of occurrence of the nucleotide in that position. The analysis was performed with sequences from 80 rescued plasmids, each one obtained from independent fungal transgenic strains. The RB sequence is over-lined. Positions 1–5 correspond to recovered gDNA sequences
In the monokaryon, similarly to the Laccaria dikaryon, the T-DNA integrations took place predominantly in the sequences annotated as putative genes (75.9 %, 66/87), while the minority of the integrations happened within intergenic regions (24.1 %, 21/87). When the sequences of the 66 interrupted genes were examined in more detail, we found that 22.7 % had integration sites in the upstream region (15/66), 39.4 % in an exon (26/66), 13.6 % in an intronic sequence (9/66) and 24.2 % (16/66) in the downstream region. Only nine of these genes had no known homologs in other organisms and half of them had a predicted associated function or encoded a conserved functional protein domain (Table 1).
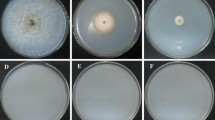
The entire collection of 1250 strains was used in growth assays on solid P5 and GPY media, out of which 13 are shown in Fig. 4. The diameters of the resulting colonies, as well as the shape of the growth borders, were compared with those obtained from the wt strain S238 N-H82. A total of 34 transgenic strains were selected based on a slightly differential growth pattern on one or both of the solid media.

Phenotypes of some L. bicolor S238 N-H82 monokaryotic T-DNA tagged strains on solid media. I GPY medium. II P5 medium. wt wild type strain. The numbers above and below the photos refer to the ID number of the transgenic strain
To further explore the potential of AMT as a functional genetics tool for L. bicolor, these selected monokaryons were mated with the compatible strain S238 N-H107 wt. The resulting dikaryons were then used in growth assays in liquid as well as on solid P5 or GPY media. Two dikaryons were found to have similar growth characteristics as the parental transgenic H82 monokaryon (strains 151 and 903; Fig. 5). In the case of the monokaryon 151, the difference in growth was originally observed as an alteration in mycelial morphology on solid medium as well as a slightly diminished growth. However, dry weight had no significant variation from the values attained for the wild type monokaryon. On the contrary, the dikaryon obtained with strain 151 had a markedly diminished dry weight when compared to the wild type dikaryon and still a slight difference when growing on solid media. For the monokaryon 903, a difference could be detected in the final dry weight in liquid media grown mycelia but not for solid media cultures. Surprisingly, the dikaryon showed a decreased growth capacity in both liquid and solid media.

Growth assays of the transgenic monokaryotic strains 151 and 903 of L. bicolor S238 N-H82 and the dikaryons resulting from the mating with strain S238 N-H107 wt. a Growth of wt and 151 strains on solid GPY medium. b Growth of wt and 903 strains on solid P5 medium. c Growth of wt and 151 strains in liquid GPY. d Growth of wt and 903 strains in liquid GPY
Discussion
In this study, we employed the previously constructed binary vector pHg/pBks for the rescue of RB–gDNA junctions in the monokaryotic strain S238 N-H82 of the basidiomycete L. bicolor after transformation by A. tumefaciens. We had already successfully employed this vector system for a T-DNA integration pattern analysis in a dikaryotic strain of the same fungus (Kemppainen et al. 2008). Those analyses demonstrated a high conservation of the RB during the integration of the T-DNA in the L. bicolor genome (Fig. 3), confirming that the RB is a good target for the T-DNA–gDNA plasmid rescue. However, if this approach should fail, the plasmid pHg/pBks has the elements required for the rescue of the LB with several restriction enzymes as well as for the simultaneous rescue of both borders by means of restriction enzymes without recognition sites within the T-DNA.
With the RB plasmid recue, we obtained positive results for the rescue of 63 % of the transgenic monokaryotic strains analyzed. Out of these, 79 % could be sequenced with the POST-RB primer, which represents 50.5 % of the total of rescues attempted. The absolute majority of these had a highly conserved RB, although several showed truncations of up to five bases. This phenomenon has been reported for plants and other fungi transformed by AMT (Oosumi et al. 2010; Maruthachalam et al. 2011; Kemski et al. 2013; Yu et al. 2015). The genomic integration sites of the rescued transgenic strains had no evident homology with the RB or LB sequences. Also, no nucleotide conservation could be found among the different integration sites, not even at single positions, microdomains or AT/CG biases.
We found that the insertion sites were located primarily in putative genes (~76 %), with nearly half of the integration events occurring within the ORFs. These results are in concordance with the findings of our previous work, where we applied the same RB rescue protocol to a dikaryotic strain of L. bicolor (Table 2). There are several models proposed for the molecular mechanisms of the T-DNA integration into the cell’s genome (for a review, see Bourras et al. 2015). A common feature of the models is the participation of multiple proteins in the process directing the insertion of the T-DNA. These proteins mostly perform the recognition of the target site (alone or in association with other cellular factors) and generally belong to the transcription regulation machinery of the transformed cell. Therefore, it is possible that the absence of a conserved sequence pattern is a consequence of the involvement of different proteins, each with its particular sequence recognition. This could also explain the preference of integration in transcriptionally active genes rather than in intergenic regions, since the T-DNA interaction partners would rarely associate with the latter. Similar studies for other fungi found that integration of the T-DNA preferentially occurred in gene-rich regions and transcriptionally active loci (Bourras et al. 2012; Kemski et al. 2013). This could be interpreted as a further support for the specific role of T-DNA integration proteins in directing the transgenes into transcriptionally active sites. Alternatively, the detected integration site bias can also be a simple artifact, a result from the selection conditions used in the isolation of the transgenic strains. As the T-DNA carries the genetic element, the transcriptional activity of which is a pre-requirement for survival and further growth of the transformed cells under selection conditions, the T-DNA integrations in euchromatic regions of the chromosomes with high transcriptional activity could be expected to allow a higher fungal survival than the heterochromatic integrations events. The results of the genome-wide T-DNA integration analysis of Arabidopsis thaliana under non-selective conditions strongly support this T-DNA integration bias (Kim and Veena 2007).
Another interesting observation is the number of integrations per genome. If the sequence data alone is regarded, there is a very high level of single integration events (more than 90 %). However, the number decreases significantly when using the Southern blot analysis (about 75 %). This difference could be due to the low number of strains employed for the latter assay when compared to the number of strains used in the rescue protocol. Also, it is possible that the transgenic strains, which produced multiple transgene signals in the Southern blot analysis, presented mixed colonies with nuclei of different T-DNA integration events. Be that as it may, the conditions employed for the AMT protocol clearly favored a single integration event per genome.
When compared to our previous results from the dikaryotic L. bicolor, we found that the percentage of successful plasmid rescue with the monokaryon was lower, even though a much larger strain set was analyzed (Table 2). On the contrary, the fraction of sequences obtained from the rescued strains was somewhat higher in the present study. The relative number of unresolved sequences remained practically identical. These correspond to strains where the integration site of the T-DNA is located very closely to a SacI recognition site or within a transposable element. In both cases, the recovered sequence is insufficient to allow the identification of a unique insertion site in the genome. Nevertheless, it is noteworthy that three out of the five insertions in transposable elements occurred within the putative coding region of the reverse transcriptase. This is in keeping with the general tendency of the T-DNA integration in gene-rich regions.
The percentage of the intergenic integrations did not vary between the AMT of monokaryotic or dikaryotic strains. Our results point to a highly reproducible outcome when using AMT on different L. bicolor genetic backgrounds and demonstrate the great potentiality of this genome manipulation technique. The starting material for a particular AMT-mediated functional genomics assay will, therefore, be determined not by the limitations of the gene transfer technique but by the desired information. As it does not require the preparation of special forms of fungal cells and employs easily cultivable mycelium, there are almost no restrictions for the use of AMT in L. bicolor functional genomics.
Another important point is the possibility of mating transgenic strains with a compatible monokaryon. In general, the dikaryotization reverted the altered growth phenotypes of the transgenic monokaryons, which indicates that the gene function driven from the second nucleus is sufficient for compensating the genetic defect of the T-DNA tagged nucleus. However, we identified two monokaryotic strains that conserved a differential growth rate even after dikaryotization. This phenomenon will have to be studied in more detail, since there are several possible explanations. One option is that the T-DNA integration results in a truncation of the gene and not its complete elimination. This could yield a partially active protein resulting in a dominant phenotype. Another option could be through gene silencing, if the truncated messenger molecule is recognized as an RNA-silencing trigger. In this case, the messengers from the second gene copy could also be affected. The realm of possibilities is wide and the only way to narrow it is through further studies.
The use of monokaryons in ectomycorrhiza studies is very limited. Normally, the dikaryotic form is the one establishing symbiosis with the plant. However, there are some reports of successful mycorrhizations with monokaryons for Hebeloma cylindrosporum (Gay et al. 1994) and Pisolithus spp. (Lamhamedi et al. 1990; Costa et al. 2010). The dikaryotization of two compatible transgenic monokaryons would allow the creation of strains defective in more than one gene of choice or in both copies of the same gene.
We have now used AMT on monokaryotic and dikaryotic strains for insertional mutagenesis, protein overexpression and gene downregulation. These studies have demonstrated some of the possible uses of this gene transfer tool in functional genomics of L. bicolor. Such great dynamic potential is necessary when attempting to unravel the highly complex genetic mechanisms that control the establishment and function of the ectomycorrhizal symbiosis.
References
Aleksenko A, Clutterbuck AJ (1997) Autonomous plasmid replication in Aspergillus nidulans: AMA1 and MATE elements. Fungal Genet Biol 21:373–387
Alonso JM, Stepanova AN, Leisse TJ, Kim CJ, Chen H, Shinn P, Stevenson DK, Zimmerman J, Barajas P, Cheuk R, Gadrinab C, Heller C, Jeske A, Koesema E, Meyers CC, Parker H, Prednis L, Ansari Y, Choy N, Deen H, Geralt M, Hazari N, Hom E, Karnes M, Mulholland C, Ndubaku R, Schmidt I, Guzman P, Aguilar-Henonin L, Schmid M, Weigel D, Carter DE, Marchand T, Risseeuw E, Brogden D, Zeko A, Crosby WL, Berry CC, Ecker JR (2003) Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 301:653–657
Aragona M, Valente MT (2015) Genetic transformation of the tomato pathogen Pyrenochaeta lycopersici allowed gene knockout using a split marker approach. Curr Genet 61:211–220
Bourras S, Meyer M, Grandaubert J, Lapalu N, Fudal I, Linglin J, Ollivier B, Blaise F, Balesdent M-H, Rouxel T (2012) Incidence of genome structure, DNA asymmetry, and cell physiology on T-DNA integration in chromosomes of the phytopathogenic fungus Leptosphaeria maculans. G3 (Bethesda) 2:891–904
Bourras S, Rouxel T, Meyer M (2015) Agrobacterium tumefaciens gene transfer: How a plant pathogen hacks the nuclei of plant and non-plant organisms. Phytopathology 105:1288–1301
Bundock P, den Dulk-Ras A, Beijersbergen A, Hooykaas PJ (1995) Trans-kingdom T-DNA transfer from Agrobacterium tumefaciens to Saccharomyces cerevisiae. EMBO J 14:3206–3214
Carvalho ND, Arentshorst M, Jin Kwon M, Meyer V, Ram AF (2010) Expanding the ku70 toolbox for filamentous fungi: establishment of complementation vectors and recipient strains for advanced gene analyses. Appl Microbiol Biotechnol 87:1463–1473
Choi J, Park J, Jeon J, Chi M-H, Goh J, Yoo S-Y, Park J, Jung K, Kim H, Park S-Y, Rho H-S, Kim S, Kim BR, Han S-S, Kang S, Lee Y-H (2007) Genome-wide analysis of T-DNA integration into the chromosomes of Magnaporthe oryzae. Mol Microbiol 66:371–382
Costa MD, Narvaes Campos, da Rocha A, Santos ML, Borges AC (2010) In vitro ectomycorrhiza formation by monokaryotic and dikaryotic isolates of Pisolithus microcarpus in Eucalyptus grandis. Revista Árvore 34:377–387
Di Battista C, Selosse M-A, Bouchard D, Stenström E, Le Tacon F (1996) Variations in symbiotic efficiency, phenotypic characters and ploidy level among different isolates of the ectomycorrhizal basidiomycete Laccaria bicolor strain S 238. Mycol Res 100:1315–1324
Gay G, Normand L, Marmeisse R, Sotta B, Debaud JC (1994) Auxin overproducer mutants of Hebeloma cylindrosporum Romagnesi have increased mycorrhizal activity. New Phytol 128:645–657
Hanif M, Pardo AG, Gorfer M, Raudaskoski M (2002) T-DNA transfer and integration in the ectomycorrhizal fungus Suillus bovinus using hygromycin B as a selectable marker. Curr Genet 41:183–188
Kemppainen M, Pardo AG (2010) pHg/pSILBAγ vector system for efficient gene silencing in homobasidiomycetes: optimization of ihpRNA–triggering in the mycorrhizal fungus Laccaria bicolor. Microb Biotechnol 3:178–200
Kemppainen M, Pardo AG (2013) LbNrt RNA silencing in the mycorrhizal symbiont Laccaria bicolor reveals a nitrate-independent regulatory role for a eukaryotic NRT2-type nitrate transporter. Environ Microbiol Rep 5:353–366
Kemppainen M, Circosta A, Tagu D, Martin F, Pardo AG (2005) Agrobacterium-mediated transformation of the ectomycorrhizal symbiont Laccaria bicolor S238 N. Mycorrhiza 16:19–22
Kemppainen M, Duplessis S, Martin F, Pardo AG (2008) T-DNA insertion, plasmid rescue and integration analysis in the model mycorrhizal fungus Laccaria bicolor. Microb Biotechnol 1:258–269
Kemppainen M, Duplessis S, Martin F, Pardo AG (2009) RNA silencing in the model mycorrhizal fungus Laccaria bicolor: gene knock-down of nitrate reductase results in inhibition of symbiosis with Populus. Environ Microbiol 11:1878–1896
Kemski MM, Stevens B, Rappleye CA (2013) Spectrum of T-DNA integrations for insertional mutagenesis of Histoplasma capsulatum. Fungal Biol 117:41–51
Kim SI, Veena Gelvin SB (2007) Genome-wide analysis of Agrobacterium T-DNA integration sites in the Arabidopsis genome generated under non-selective conditions. Plant J 51:779–791
Kim JK, Park YJ, Kong WS, Kang HW (2010) Highly efficient electroporation-mediated transformation into edible mushroom Flammulina velutipes. Mycobiology 38:331–335
Klein T, Siegwolf RT, Körner C (2016) Belowground carbon trade among tall trees in a temperate forest. Science 352:342–344
Kuo C-Y, Huang C-T (2008) A reliable transformation method and heterologous expression of beta-glucuronidase in Lentinula edodes. J Microbiol Methods 72:111–115
Lamhamedi MS, Fortin JA, Kope HH, Kropp BR (1990) Genetic variation in ectomycorrhiza formation by Pisolithus arhizus on Pinus pinaster and Pinus banksiana. New Phytol 115:689–697
Li G, Li R, Liu Q, Wang Q, Chen M, Li B (2006) A highly efficient polyethylene glycol-mediated transformation method for mushrooms. FEMS Microbiol Lett 256:203–208
Lo Presti L, Lanver D, Schweizer G, Tanaka S, Liang L, Tollot M, Zuccaro A, Reissmann S, Kahmann R (2015) Fungal effectors and plant susceptibility. Annu Rev Plant Biol 66:513–545
Lucic E, Fourrey C, Kohler A, Martin F, Chalot M, Brun-Jacob A (2008) A gene repertoire for nitrogen transporters in Laccaria bicolor. New Phytol 180:343–364
Martin F, Aerts A, Ahrén D, Brun A, Danchin EGJ, Duchaussoy F, Gibon J, Kohler A, Lindquist E, Pereda V, Salamov A, Shapiro HJ, Wuyts J, Blaudez D, Buée M, Brokstein P, Canbäck B, Cohen D, Courty PE, Coutinho PM, Delaruelle C, Detter JC, Deveau A, DiFazio S, Duplessis S, Fraissinet-Tachet L, Lucic E, Frey-Klett P, Fourrey C, Feussner I, Gay G, Grimwood J, Hoegger PJ, Jain P, Kilaru S, Labbé J, Lin YC, Legué V, Le Tacon F, Marmeisse R, Melayah D, Montanini B, Muratet M, Nehls U, Niculita-Hirzel H, Secq MPO-L, Peter M, Quesneville H, Rajashekar B, Reich M, Rouhier N, Schmutz J, Yin T, Chalot M, Henrissat B, Kües U, Lucas S, Van de Peer Y, Podila GK, Polle A, Pukkila PJ, Richardson PM, Rouzé P, Sanders IR, Stajich JE, Tunlid A, Tuskan G, Grigoriev IV (2008) The genome of Laccaria bicolor provides insights into mycorrhizal symbiosis. Nature 452:88–92
Maruthachalam K, Klosterman SJ, Kang S, Hayes RJ, Subbarao KV (2011) Identification of pathogenicity-related genes in the vascular wilt fungus Verticillium dahliae by Agrobacterium tumefaciens-mediated T-DNA insertional mutagenesis. Mol Biotechnol 49:209–221
Müller T, Benjdia M, Avolio M, Voigt B, Menzel D, Pardo A, Frommer WB, Wipf D (2006) Functional expression of the green fluorescent protein in the ectomycorrhizal model fungus Hebeloma cylindrosporum. Mycorrhiza 16:437–442
Navarro-Ródenas A, Xu H, Kemppainen M, Pardo AG, Zwiazek JJ (2015) Laccaria bicolor aquaporin LbAQP1 is required for Hartig net development in trembling aspen (Populus tremuloides). Plant Cell Environ 38:2475–2486
Nygren CMR, Eberhardt U, Karlsson M, Parrent JL, Lindahl BD, Taylor AFS (2008) Growth on nitrate and occurrence of nitrate reductase-encoding genes in a phylogenetically diverse range of ectomycorrhizal fungi. New Phytol 180:875–889
Oosumi T, Ruiz-Rojas JJ, Veilleux RE, Dickerman A, Shulaev V (2010) Implementing reverse genetics in Rosaceae: analysis of T-DNA flanking sequences of insertional mutant lines in the diploid strawberry, Fragaria vesca. Physiol Plant 140:1–9
Pardo AG, Hanif M, Raudaskoski M, Gorfer M (2002) Genetic transformation of ectomycorrhizal fungi mediated by Agrobacterium tumefaciens. Mycol Res 106:132–137
Peterson L, Massicotte HB, Melville LH (2004) Mycorrhizas: anatomy and cell biology. NCR Research Press, Otawa
Piers KL, Heath JD, Liang X, Stephens KM, Nester EW (1996) Agrobacterium tumefaciens-mediated transformation of yeast. Proc Natl Acad Sci USA 93:1613–1618
Plett JM, Kemppainen M, Kale SD, Kohler A, Legué V, Brun A, Tyler BM, Pardo AG, Martin F (2011) A secreted effector protein of Laccaria bicolor is required for symbiosis development. Curr Biol 21:1197–1203
Plett JM, Khachane A, Ouassou M, Sundberg B, Kohler A, Martin F (2014a) Ethylene and jasmonic acid act as negative modulators during mutualistic symbiosis between Laccaria bicolor and Populus roots. New Phytol 202:270–286
Plett JM, Daguerre Y, Wittulsky S, Vayssières A, Deveau A, Melton SJ, Kohler A, Morrell-Falvey JL, Brun A, Veneault-Fourrey C, Martin F (2014b) Effector MiSSP7 of the mutualistic fungus Laccaria bicolor stabilizes the Populus JAZ6 protein and represses jasmonic acid (JA) responsive genes. Proc Natl Acad Sci USA 111:8299–8304
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning. A laboratory Manual, 2nd edn, Cold Spring Harbor Laboratory Press, New York
Schneeberger RG, Zhang K, Tatarinova T, Troukhan M, Kwok SF, Drais J, Klinger K, Orejudos F, Macy K, Bhakta A, Burns J, Subramanian G, Donson J, Flavell R, Feldmann KA (2005) Agrobacterium T-DNA integration in Arabidopsis is correlated with DNA sequence compositions that occur frequently in gene promoter regions. Funct Integr Genomics 5:240–253
Selosse MA, Costa G, Battista CD, Tacon FL, Martin F (1996) Meiotic segregation and recombination of the intergenic spacer of the ribosomal DNA in the ectomycorrhizal basidiomycete Laccaria bicolor. Curr Genet 30:332–337
Shi L, Fang X, Li M, Mu D, Ren A, Tan Q, Zhao M (2012) Development of a simple and efficient transformation system for the basidiomycetous medicinal fungus Ganoderma lucidum. World J Microbiol Biotechnol 28:283–291
Smith SE, Read D (2008) Mycorrhizal symbiosis. Academic Press, London
Tatry M-V, El Kassis E, Lambilliotte R, Corratgé C, van Aarle I, Amenc LK, Alary R, Zimmermann S, Sentenac H, Plassard C (2009) Two differentially regulated phosphate transporters from the symbiotic fungus Hebeloma cylindrosporum and phosphorus acquisition by ectomycorrhizal Pinus pinaster. Plant J 57:1092–1102
Wright DP, Johansson T, Le Quéré A, Söderström B, Tunlid A (2005) Spatial patterns of gene expression in the extramatrical mycelium and mycorrhizal root tips formed by the ectomycorrhizal fungus Paxillus involutus in association with birch (Betula pendula) seedlings in soil microcosms. New Phytol 167:579–596
Xu H, Kemppainen M, El Kayal W, Lee SH, Pardo AG, Cooke JEK, Zwiazek JJ (2015) Overexpression of Laccaria bicolor aquaporin JQ585595 alters root water transport properties in ectomycorrhizal white spruce (Picea glauca) seedlings. New Phytol 205:757–770
Xu H, Cooke JEK, Kemppainen M, Pardo AG, Zwiazek JJ (2016) Hydraulic conductivity and aquaporin transcription in roots of trembling aspen (Populus tremuloides) seedlings colonized by Laccaria bicolor. Mycorrhiza, p 1–11
Yin Y, Liu Y, Jin H, Wang S, Zhao S, Geng X, Li M, Xu F (2012) Polyethylene glycol-mediated transformation of fused egfp-hph gene under the control of gpd promoter in Pleurotus eryngii. Biotechnol Lett 34:1895–1900
Yu M, Yu J, Hu J, Huang L, Wang Y, Yin X, Nie Y, Meng X, Wang W, Liu Y (2015) Identification of pathogenicity-related genes in the rice pathogen Ustilaginoidea virens through random insertional mutagenesis. Fungal Genet Biol 76:10–19
Zhang J, Guo D, Chang Y, You C, Li X, Dai X, Weng Q, Zhang J, Chen G, Li X, Liu H, Han B, Zhang Q, Wu C (2007) Non-random distribution of T-DNA insertions at various levels of the genome hierarchy as revealed by analyzing 13 804 T-DNA flanking sequences from an enhancer-trap mutant library. Plant J 49:947–959
Zhang JJ, Shi L, Chen H, Sun YQ, Zhao MW, Ren A, Chen MJ, Wang H, Feng ZY (2014) An efficient Agrobacterium-mediated transformation method for the edible mushroom Hypsizygus marmoreus. Microbiol Res 169:741–748
Zubieta MP, da Silva Coelho I, Queiroz MV, Araújo EF (2014) Agrobacterium tumefaciens-mediated genetic transformation of the ectomycorrhizal fungus Laccaria laccata. Ann Microbiol 64:1875–1878
Acknowledgments
The authors wish to thank UNQ, CONICET and ANPCyT for financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Kupiec.
B. I. Stephan and M. C. Alvarez Crespo contributed equally to this work.
Rights and permissions
About this article

Cite this article
Stephan, B.I., Alvarez Crespo, M.C., Kemppainen, M.J. et al. Agrobacterium-mediated insertional mutagenesis in the mycorrhizal fungus Laccaria bicolor . Curr Genet 63, 215–227 (2017). https://doi.org/10.1007/s00294-016-0627-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-016-0627-x