
Abstract
We sequenced the complete plastid and mitochondrial genomes of the unicellular marine phytoplankton Triparma laevis, belonging to the order Parmales (Heterokonta). The cells of Parmales are surrounded by silicified cell walls, similar to Bacillariophyta (diatoms). T. laevis was recognized as a sister group of Bacillariophyta using a molecular phylogenetic analysis based on SSU rDNA and rbcL sequences. Bacillariophyta are the most successful group of phytoplankton in the modern ocean, but the origin and early evolution of them have not been clearly established. Detailed molecular analyses of T. laevis may increase our understanding of the evolutionary relationships among Parmales and Bacillariophyta. The gene contents of the plastid and mitochondrial genomes are similar between T. laevis and Bacillariophyta. The gene order of the plastid genome is also similar to Bacillariophyta, whereas the gene order of the mitochondrial genome is not conserved in Bacillariophyta, but the structure is more compact than Bacillariophyta. Phylogenetic analyses, using plastid-encoded concatenated amino acid datasets and mitochondria-encoded concatenated amino acid datasets suggest that T. laevis is a sister group of Bacillariophyta. These results suggest that the characteristics of the organellar genomes of T. laevis are similar and conserve ancestral characteristics more than Bacillariophyta.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Plastids and mitochondria are organelles that have their own genomes and genetic systems. Endosymbiotic cyanobacteria and alphaproteobacteria are believed to be the origin of plastids and mitochondria, respectively (Sagan 1967; Williams et al. 2007). Plastids are classified by origin; one is a direct descendent of a primary cyanobacterium endosymbiont, while the other originated from the endosymbiosis of plastid-bearing eukaryotes by another eukaryotic host (Kim and Archibald 2009). The plastids of Heterokonta originated from Rhodophyta (Cavalier-Smith 1998; Tajima et al. 2014). The mitochondria of Heterokonta originated from eukaryotic hosts of secondary plastids (Prihoda et al. 2012). Comprehensive and comparative organelle genome sequence analyses will improve our understanding of the evolutionary relationships between Parmales and other Heterokonta.
The order Parmales (Heterokonta) is a group of unicellular marine phytoplankton species with small solitary cells that are generally about 2–5 µm in diameter (Booth and Marchant 1987). The cells are surrounded by variously shaped plates composed of silica (Silver et al. 1980). Their taxonomic status was based on the structure and number of plates of the siliceous cell wall in samples collected from natural habitats (Booth and Marchant 1987; Kosman et al. 1993). The order Parmales is divided into two families, Triparmaceae (including Tetraparma and Triparma) and Pentalaminaceae. Parmales is distributed globally, from tropical to polar waters; however, its abundance is particularly high in polar and subarctic waters (Booth et al. 1980; Nishida 1986; Komuro et al. 2005; Konno and Jordan 2007; Ichinomiya and Kuwata 2015).
Parmalean algae have never been cultured successfully because they are small and difficult to distinguish from other small phytoplankton in field samples by light microscopy. Recently, using the silicified cell wall staining technique, a parmalean culture (Triparma laevis strain NIES-2565) was successfully established in the Oyashio region of the western North Pacific (Ichinomiya et al. 2011). Tentatively, Parmales belong to the class Chrysophyceae (Booth and Marchant 1987), but the molecular phylogenetic analyses based on 18S ribosomal DNA and rbcL sequences indicated that Triparma is not included within the Chrysophyceae, but instead is the sister group of Bacillariophyta (diatoms) (Ichinomiya et al. 2011).
Diatoms are the most successful group of phytoplankton in the modern ocean; they include ca. 105 species (Mann and Vanormelingen 2013) and have high primary productivity, which is estimated to contribute about 20 % of the photosynthesis on earth, comparable to that of terrestrial rainforests (Nelson et al. 1995; Field et al. 1998; Mann 1999). Because the origin and early evolution of diatoms have not been clearly established, Parmales studies may play a key role in understanding the early evolution of Bacillariophyta.
Here, we report the sequence of the plastid and mitochondrial genomes of T. laevis strain NIES-2565. We analyzed their genome structures and gene contents, and compared their characteristics with those of Bacillariophyta and other Heterokonta. The organellar genomes of some species of Heterokonta have been sequenced and compared gene contents and gene order between the related species (Starkenburg et al. 2014). The gene repertoires and gene order of both organelle genomes of Bacillariophyta are similar (Oudot-Le Secq and Green 2011; Oudot-Le Secq et al. 2007; Ruck et al. 2014). Comparative analyses of organelle genomes will improve our understanding of the similarity and evolutionary relationships between Parmales and Bacillariophyta. In addition, using concatenated amino acid alignments of plastid-encoded proteins and mitochondria-encoded proteins, we estimated the phylogenetic relationship between T. laevis and other Heterokonta.
Materials and methods
DNA sources
Triparma laevis strain NIES-2565 was isolated from the Oyashio region of the western North Pacific. Cells of T. laevis were maintained in modified f/2 medium (Guillard and Ryther 1962) with 100 µM nitrate and 100 µM silicic acid in a plastic bottle at 5 °C under 100 µmol photons m−2 s−1 (14 h of light: 10 h of dark) (Ichinomiya et al. 2011). The cells were harvested by filtration. DNA was released by treatment with sodium N-dodecanoylsarcosinate and proteinase K, extracted with phenol/chloroform, and purified by CsCl ultracentrifugation.
DNA sequencing
Genomic DNA (0.5 µg for shotgun and 5 µg for paired-end libraries) was sheared and sequenced with a genome sequencer 454 FLX+ (Roche Diagnostics, Indianapolis, IN, USA) according to the manufacturer’s protocol. Total genome data of T. laevis were assembled with Newbler version 2.6 into 12,625 contigs (average length = 4647 bp). The contigs had read depth of 22.5 on an average. To determine the genomic origin (i.e., nuclear genome or organellar genomes), each contig was analyzed by blastn version 2.2.18 (Altschul et al. 1997) against the sequences of the organellar and nuclear genomes in algae and plants. The sequence reads corresponding to plastid sequences were assembled into 21 contigs and a single mitochondrial scaffold. PCR experiments were performed to fill the gaps. The products were sequenced by conventional Sanger sequencing by FASMAC Co. Ltd. (Atsugi, Japan). The complete organellar genomes are available under GenBank Accession Numbers AP014625 (plastid genome) and AP014626 (mitochondrial genome), respectively.
Genome annotation and data analyses
To detect the open reading frames (ORFs) of the plastid genome, we used the MetaGeneAnnotator program (Noguchi et al. 2008). We then used the tblastn program, which was run against the cluster data of CyanoClust (Sasaki and Sato 2010) prepared by Gclust (Sato 2009) (Supplementary material 2). Finally, we searched for ORFs that were longer than 30 codons and started with ATG or GTG. To detect the ORFs of the mitochondrial genome, we used MetaGeneAnnotator. We then used tblastn, which was run against the cluster data of 23 mitochondrial genomes (Dataset: Mt23) prepared by Gclust (Supplementary material 2). To search for tRNA genes, we used the program tRNAscan-SE (Lowe and Eddy 1997). To detect rRNA genes, we used the blastn program against the plastid genomes of Rhodophyta and Glaucophyta and the mitochondrial genomes of the species in the Mt23 dataset (Supplementary material 2). The gene orders of two non-coding RNAs (ncRNAs; ffs and ssrA) were conserved in Bacillariophyta (ffs is between tRNAPhe(GAA) and psbX and ssrA is between tRNAArg(CCG) and tRNAIle(CAU)). We then constructed a sequence alignment between tRNAPhe(GAA) and psbX of T. laevis and ffs genes of Bacillariophyta. We also constructed a sequence alignment between tRNAArg(CCG) and tRNAIle(CAU) of T. laevis and ssrA genes of Bacillariophyta. We hypothesized that the highly homologous regions encoded ncRNA genes in T. laevis. Processing of DNA and protein sequences was performed with SISEQ version 1.59 (Sato 2000). A sequence alignment of ncRNA genes was constructed with Clustal X version 2.0.9 (Thompson et al. 1994; Larkin et al. 2007). The genomic sequence was manipulated using Artemis version 13.0 (Rutherford et al. 2000).
Phylogenetic analyses
For phylogenetic analyses of Heterokonta based on the plastid genomes, two types of protein sequence alignments were constructed: one type used sequences that were conserved in five species of Rhodophyta, six species of Viridiplantae, one species of Glaucophyta, 19 species of Heterokonta, three species of Haptophyta, two species of Cryptophyta, and 18 species of cyanobacteria, and another type used sequences conserved in five species of Rhodophyta, 19 species of Heterokonta, three species of Haptophyta, two species of Cryptophyta, and 18 species of cyanobacteria. For phylogenetic analyses of Heterokonta based on mitochondrial genomes, four types of protein sequence alignments were constructed: three types used sequences conserved in three species of Rhodophyta, six species of Viridiplantae, two species of Glaucophyta, 13 species of Heterokonta, five species of alphaproteobacteria, and 0, 1, or 2 species of Haptophyta and Cryptophyta, respectively, and another type used sequences conserved in 13 species of Heterokonta and five species of alphaproteobacteria. The analyzed proteins are listed in Supplementary material 3. The alignments of concatenated protein sequences are described in Supplementary material 4–9. We used the ML (maximum likelihood) and BI (Bayesian inference) methods for phylogenetic inference. An ML analysis was performed using PhyML version 3.1 (Guindon et al. 2010). The evolutionary model was selected with ProtTest version 2.4 (Abascal et al. 2005). The LG (Le and Gascuel 2008) model was selected, but because it was not implemented in MrBayes, the best available model was used. A BI analysis was performed using MrBayes version 3.2.1 (Ronquist and Huelsenbeck 2003) with the WAG (Whelan and Goldman 2001) model. Two chains were run; trees were sampled at every 200 for 1,000,000 generations and the first 2,000 trees were discarded as burn-in.
Phylogenetic analyses of PsbA
For phylogenetic analyses of PsbA, we used sequences in five species of Rhodophyta, 11 species of Heterokonta, one species of Haptophyta, and two species of Cryptophyta. The phylogenetic relationship was analyzed using the ML method with the LG model. The alignment of PsbA is provided in Supplementary material 10. To compare the sequences of the two copies of PsbA in T. laevis, a sequence alignment was constructed with Clustal X software version 2.0.9.
Results
Structure of the organellar genomes of T. laevis
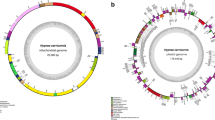
We sequenced the plastid and mitochondrial genomes of T. laevis strain NIES-2565 by pyrosequencing with the Roche 454 GS FLX system. The sequence reads of the plastid genome were assembled into 21 contigs and the gaps were filled completely by PCR and Sanger sequencing. The plastid genome of T. laevis is a circular molecule composed of 117,514 bp, with a GC content of 31.7 %, and encodes two ncRNAs (ffs and ssrA), six rRNAs (two rrs, two rrl, and two rrf), 29 tRNAs, and 141 proteins (Fig. 1a, Table S1, and Supplementary material 12). This plastid genome encodes two copies of psbA, which are different in coding sequence but phylogenetically similar (Fig. S1). These results suggest that the psbA genes underwent duplication and mutation after the diversification of Parmales from Bacillariophyta. Sequence reads of the mitochondrial genome were assembled into one scaffold, and the gaps were filled completely by PCR and Sanger sequencing. The mitochondrial genome of T. laevis is a circular molecule composed of 39,580 bp with a GC content of 30.4 %, and encodes two rRNAs (rrs and rrl), 25 tRNAs, and 37 proteins (Fig. 1b, Table S3, and Supplementary material 12). The number of uaa stop codons was 35, whereas there were only two uag stop codons. The uga codon, used as a stop codon by many species and as a Trp codon by some species (Fox 1979; Oudot-Le Secq and Green 2011), was not used as a stop or Trp codon in the mitochondrial genome of T. laevis (Table S4).

Gene maps of the organellar genomes of T. laevis. The two internal sectors represent RNA genes, whereas the two external sectors represent protein-coding genes. The outer and inner rings of each set represent genes on the clockwise and counterclockwise strands, respectively. Protein-coding genes are color coded by their function, as shown at the bottom left corner. a Gene map of the plastid genome of T. laevis. b Gene map of the mitochondrial genome of T. laevis
Comparison of protein-coding genes in plastids
We analyzed the loss of plastid protein-coding genes during the evolution of Heterokonta (Fig. S2). A previous report showed that the plastids of Heterokonta originated from Rhodophytina (Tajima et al. 2014). The plastid genomes of Heterokonta have lost 51 genes compared to Rhodophytina. The plastid genomes of T. laevis and Bacillariophyta have lost 13 genes after diverging from other Heterokonta. The peroxiredoxin gene (bas1), which has been lost repeatedly from the plastid genomes of Rhodophyta and Bacillariophyta (Glöckner et al. 2000; Ruck et al. 2014), was lost from the plastid genome of T. laevis. The genes chlB, chlL, chlN, rpoZ, and ycf19 were lost from the plastid genomes of Bacillariophyta after diverging from T. laevis, and several genes showed lineage-specific loss (Lommer et al. 2010; Oudot-Le Secq et al. 2007; Ruck et al. 2014). The petJ gene, lost from the plastid genome of T. laevis, was detected in the draft nuclear genome of T. laevis (unpublished data). This gene appears to have been transferred to the nucleus, as in some species of Bacillariophyta (Ruck et al. 2014). No other genes were lost from the plastid genome of T. laevis after diverging from Bacillariophyta.
The plastid genomes of several species contained serC and/or tyrC, functional recombinase genes that originated from foreign species (Ruck et al. 2014). The genes ycf88, ycf89, and ycf90, conserved in only Bacillariophyta (ycf88 is conserved in Bacillariophyta, excluding Leptocylindrus danicus), may have originated from the plastid genomes of Bacillariophyta. These genes may have been acquired by lineage-specific gains.
Comparison of plastid genomes
We compared the global structure of the plastid genome of T. laevis with that of Heterokonta (Supplementary material 11). The gene order of T. laevis was similar to that of Bacillariophyta, compared with other Heterokonta. Three tRNA(CAU)s of T. laevis were identified as tRNAfMet(CAU), tRNAIle(CAU), and tRNAMet(CAU) based on an alignment analysis (Table S2) (Tajima et al. 2014). We also determined and corrected the identity of tRNA(CAU)s from Heterokonta plastid genomes that were unidentified or misannotated.
Some species of Bacillariophyta contained many duplicated genes in the inverted repeat region. The inverted repeat regions of T. laevis contained the rRNA operon and part of the hypothetical protein (ORF50 and ORF51).
Based on the repertoire and order of plastid genes, we identified roughly 18 blocks in T. laevis and Bacillariophyta. The block trnT(UGU)-trnY(GUA) was often rearranged. Two blocks of gene order were not conserved in T. laevis and other Bacillariophyta; the block psaJ-trnK(UUU) was separated in L. danicus, Thalassiosira pseudonana, and T. laevis, and rps14 was not adjacent to psaM in L. danicus, Coscinodiscus radiatus, and T. laevis.
The gene repertoire in the small single-copy region within Bacillariophyta was conserved with the exception of species-specific genes. The plastid genome of T. laevis encoded the block of the ribosomal gene cluster (dnaK-rps10) and two neighboring blocks (ilvH-clpC and groEL-rps16) in the large single-copy region.
Comparison of mitochondrial genomes
We compared the mitochondrial genome of T. laevis with that of Heterokonta. Table 1 shows the difference in gene content among lineages. The trnT gene was absent in all Heterokonta. Full-length nad11 was encoded in Raphidophyceae, Synurophyceae, Chrysophyceae, Bacillariophyta, and T. laevis. In T. laevis, the similarity of the sequence encoding the C-terminal “molybdopterin-binding” domain of nad11 was low. In some species of Bacillariophyta, nad11 was split into two ORFs. Only the N-terminal “Fe-S binding” domain of nad11 was identified in Phaeophyceae (Oudot-Le Secq and Green 2011), whereas only the C-terminal “molybdopterin-binding” domain was identified in Eustigmatophyceae (Starkenburg et al. 2014). An in-frame insertion was found in cox2 in Eustigmatophyceae and Phaeophyceae (excluding Dictyota dichotoma) (Oudot-Le Secq and Green 2011). The only difference in gene content of the mitochondrial genome between T. laevis and Bacillariophyta was that the tRNAGly(UCC) gene was lost from Bacillariophyta. The gene order of the mitochondrial genome of T. laevis was not similar to that of Bacillariophyta. All species of Bacillariophyta had introns and some species had a block of repeated sequence (Oudot-Le Secq and Green 2011), whereas T. laevis had no introns or repeat regions.
Phylogenetic analyses of Heterokonta based on organellar genomes
For phylogenetic analyses of Heterokonta based on the plastid genome, we constructed an amino acid sequence alignment of 33 concatenated proteins that were conserved in Rhodophyta, Viridiplantae, Glaucophyta, Heterokonta, Haptophyta, Cryptophyta, and cyanobacteria (Fig. 2). Additionally, we constructed an amino acid sequence alignment of 64 concatenated proteins that were conserved in Rhodophyta, Heterokonta, Haptophyta, Cryptophyta, and cyanobacteria (Fig. S3). We removed genes that may provide a misleading phylogenetic signal; namely, serially duplicated genes (psbA and psbD), genes of short sequence alignment (psbF, psbH, psbJ, psbK, psbN, psbT, rpl31, rpl33, and rps18) (Qiu et al. 2012), and horizontally transferred genes (rbcL, rbcS, and rpl36) (Delwiche and Palmer 1996; Rice and Palmer 2006). We used ML and BI methods. The results of our analyses in both datasets indicated that T. laevis is closely related to Bacillariophyta. The obtained phylogenetic relationships among Heterokonta, Haptophyta, Cryptophyta, and Rhodophyta were consistent with those of previous reports (Tajima et al. 2014).

Phylogenetic tree of plastids and cyanobacteria based on concatenated sequences of 33 conserved proteins. The bar above the tree shows the scale for the branch length (substitutions per site). The numbers at each branch indicate the confidence level and posterior probability, provided by ML (first value) and BI (second value) analyses, respectively. “−” indicates that this branch was not supported by BI analysis. The tree topology as shown here was identical in the ML analyses. The thick lines represent the branches that were completely supported by the two analyses
For phylogenetic analyses of Heterokonta based on the mitochondrial genome, we constructed an amino acid sequence alignment of 29 concatenated proteins that were conserved in Heterokonta with alphaproteobacteria as an outgroup (Fig. 3). Additionally, we constructed three amino acid sequence alignments of 15 concatenated proteins that were conserved in Rhodophyta, Viridiplantae, Glaucophyta, Heterokonta, Haptophyta, Cryptophyta, and alphaproteobacteria (Fig. S4). These datasets included 0, 1, or 2 species each of Haptophyta and Cryptophyta, respectively. The phylogenetic relationships of Heterokonta were supported in all datasets by both analyses. These results also indicated that T. laevis is closely related to Bacillariophyta. However, the phylogenetic relationships of Heterokonta with other lineages varied by dataset and analysis. The results of the datasets with or without one species each of Haptophyta and Cryptophyta suggested that Viridiplantae, Glaucophyta, and Heterokonta form a monophyletic group. On the other hand, the results of the dataset with two species each of Haptophyta and Cryptophyta suggested that Rhodophyta, Viridiplantae, Glaucophyta, Haptophyta, and Cryptophyta are a monophyletic group, excluding Heterokonta as an outgroup. These results are not consistent with the results of Oudot-Le Secq et al. (2006). These differences may be due to differences in the rate of mitochondrial mutations among diverse lineages (Drouin et al. 2008; Felsenstein 1978; Smith et al. 2012, 2014).

Phylogenetic tree of mitochondria and alphaproteobacteria based on concatenated sequences of 29 conserved proteins. The bar above the tree shows the scale for the branch length (substitutions per site). The numbers at each branch indicate the confidence level and posterior probability, provided by ML (first value) and BI (second value) analyses, respectively. The tree topology as shown here was identical in the ML analyses. The thick lines represent the branches that were supported by the two analyses (ML: = 100; BI: >99)
Discussion
We sequenced the complete plastid and mitochondrial genomes of T. laevis. The gene repertoires of both organelle genomes are similar to those of Bacillariophyta. The plastid genomes of T. laevis and Bacillariophyta do not have as many inversions as other Heterokonta. This suggests that large-scale genome rearrangement did not occur in plastid genomes in the lineages of T. laevis and Bacillariophyta after their separation from the ancestor of other Heterokonta. The gene order of the plastid genome is similar to that of Bacillariophyta, whereas the gene order of the mitochondrial genome is dissimilar to that of Bacillariophyta. Bacillariophyta have introns and a specific block of repeat sequences in each species, but the gene order in the large block containing most of the ribosomal protein genes is conserved (Oudot-Le Secq and Green 2011). This result implies that large-scale mitochondrial genome rearrangements occurred after Parmales diverged from Bacillariophyta. The organellar genomes of T. laevis implied more compact structures and encoded some genes that were lost in Bacillariophyta. These features suggest that the structure of the organellar genomes of T. laevis may conserve ancestral characteristics more than Bacillariophyta.
The molecular phylogenetic analyses based on 18S ribosomal DNA and rbcL sequences indicated that T. laevis is included within the bolidophycean clade (Ichinomiya et al. 2011). Bolidophycean algae are small, naked flagellates that have been recognized as a sister group of Bacillariophyta based on their molecular phylogeny (Guillou et al. 1999). Some plastid-encoded genes of Bolidophyceae have been partially sequenced (Daugbjerg and Guillou 2001; Yang et al. 2012); each amino acid identity of psaA gene between T. laevis and Bolidomonas pacifica CCMP 1866, Bolidomonas mediterranea CCMP 1867, L. danicus, and C. radiatus is 450/460, 442/460, 438/460, and 434/460, and each amino acid identity of rbcL gene between T. laevis and B. pacifica var. eleuthera, B. mediterranea CCMP 1867, L. danicus, and C. radiatus is 462/474, 449/474, 438/474, and 437/474. These data also suggested Bolidophyceae have closer relationship with T. laevis than Bacillariophyta.
Sequencing of the complete organellar genomes of T. laevis and comprehensive and comparative organelle genome sequence analyses increased our understanding of the evolutionary relationships between Parmales and other Heterokonta. Sequencing of the complete organellar genomes of additional species of Parmales and Bolidophyceae will reveal whether the genome structures and some genes that were lost in Bacillariophyta are conserved in Parmales and Bolidophyceae. Sequencing of the nuclear genome of T. laevis will reveal the features of the genome of T. laevis and increase our understanding of the evolution of Bacillariophyta and Parmales.
References
Abascal F, Zardoya R, Posada D (2005) ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21:2104–2105
Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402
Booth BC, Marchant HJ (1987) Parmales, a new order of marine chrysophytes, with descriptions of three new genera and seven new species. J Phycol 23:245–260
Booth BC, Lewin J, Norris RE (1980) Siliceous nanoplankton. 1. Newly discovered cysts from the Gulf of Alaska. Mar Biol 58:205–209
Cavalier-Smith T (1998) A revised six-kingdom system of life. Biol Rev Camb Philos Soc 73:203–266
Daugbjerg N, Guillou L (2001) Phylogenetic analyses of Bolidophyceae (Heterokontophyta) using rbcL gene sequences support their sister group relationship to diatoms. Phycologia 40:153–161
Delwiche CF, Palmer JD (1996) Rampant horizontal transfer and duplication of rubisco genes in eubacteria and plastids. Mol Biol Evol 13:873–882
Drouin G, Daoud H, Xia J (2008) Relative rates of synonymous substitutions in the mitochondrial, chloroplast and nuclear genomes of seed plants. Mol Phylogenet Evol 49:827–831
Felsenstein J (1978) Cases in which parsimony or compatibility methods will be positively misleading. Syst Zool 27:401–410
Field CB, Behrenfeld MJ, Randerson JT, Falkowski P (1998) Primary production of the biosphere: integrating terrestrial and oceanic components. Science 281:237–240
Fox TD (1979) Five TGA “stop” codons occur within the translated sequence of the yeast mitochondrial gene for cytochrome c oxidase subunit II. Proc Natl Acad Sci USA 76:6534–6538
Glöckner G, Rosenthal A, Valentin K (2000) The structure and gene repertoire of an ancient red algal plastid genome. J Mol Evol 51:382–390
Guillard RRL, Ryther JH (1962) Studies of marine planktonic diatoms I. Cyclotella nana Hustedt and Detonula confervacea (cleve) Gran. Can J Microbiol 8:229–239
Guillou L, Chrétiennot-Dinet M-J, Medlin LK, Claustre H, Loiseaux-de Göer S, Vaulot D (1999) Bolidomonas: a new genus with two species belonging to a new algal class, the Bolidophyceae (Heterokonta). J Phycol 35:368–381
Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, Gascuel O (2010) New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59:307–321
Ichinomiya M, Kuwata A (2015) Seasonal variation in abundance and species composition of the Parmales community in the Oyashio region, western North Pacific. Aquat Microb Ecol 75:207–223
Ichinomiya M, Yoshikawa S, Kamiya M, Ohki K, Takaichi S, Kuwata A (2011) Isolation and characterization of Palmales (Heterokonta/Heterokontophyta/Stramenopiles) from the Oyashio region, western North Pacific. J Phycol 47:144–151
Kim E, Archibald JM (2009) Diversity and evolution of plastids and their genomes. The chloroplast—interactions with the environment. In: Aronsson H, Sandelius AS (eds) Plant cell monographs, vol 13. Springer, Berlin, pp 1–39
Komuro C, Narita H, Imai K, Nojiri Y, Jordan RW (2005) Microplankton assemblages at Station KNOT in the subarctic western Pacific, 1999–2000. Deep Sea Res Part II: Top Stud Oceanogr 52:2206–2217
Konno S, Jordan RW (2007) An amended terminology for the Parmales (Chrysophyceae). Phycologia 46:612–616
Kosman CA, Thomsen HA, Østergaard JB (1993) Parmales (Chrysophyceae) from Mexican, Californian, Baltic, Arctic and Antarctic waters with the description of a new subspecies and several new forms. Phycologia 32:116–128
Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R et al (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948
Le SQ, Gascuel O (2008) An improved general amino acid replacement matrix. Mol Biol Evol 25:1307–1320
Lommer M, Roy AS, Schilhabel M, Schreiber S, Rosenstiel P, LaRoche J (2010) Recent transfer of an iron-regulated gene from the plastid to the nuclear genome in an oceanic diatom adapted to chronic iron limitation. BMC Genom 11:718
Lowe TM, Eddy SR (1997) tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25:955–964
Mann DG (1999) The species concept in diatoms. Phycologia 38:437–495
Mann DG, Vanormelingen P (2013) An inordinate fondness? The number, distributions, and origins of diatom species. J Eukaryot Microbiol 60:414–420
Nelson DM, Tréguer P, Brzezinski MA, Leynaert A, Quéguiner B (1995) Production and dissolution of biogenic silica in the ocean: revised global estimates, comparison with regional data and relationship to biogenic sedimentation. Global Biogeochem Cycles 9:359–372
Nishida S (1986) Nanoplankton flora in the Southern Ocean, with special reference to siliceous varieties. Mem Nat Inst Polar Res Spec Issue 40:56–68
Noguchi H, Taniguchi T, Itoh T (2008) MetaGeneAnnotator: detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res 15:387–396
Oudot-Le Secq MP, Green BR (2011) Complex repeat structures and novel features in the mitochondrial genomes of the diatoms Phaeodactylum tricornutum and Thalassiosira pseudonana. Gene 476:20–26
Oudot-Le Secq MP, Loiseaux-de Goër S, Stam WT, Olsen JL (2006) Complete mitochondrial genomes of the three brown algae (Heterokonta: Phaeophyceae) Dictyota dichotoma, Fucus vesiculosus and Desmarestia viridis. Curr Genet 49:47–58
Oudot-Le Secq MP, Grimwood J, Shapiro H, Armbrust EV, Bowler C, Green BR (2007) Chloroplast genomes of the diatoms Phaeodactylum tricornutum and Thalassiosira pseudonana: comparison with other plastid genomes of the red lineage. Mol Genet Genomics 277:427–439
Prihoda J, Tanaka A, de Paula WB, Allen JF, Tirichine L, Bowler C (2012) Chloroplast-mitochondria cross-talk in diatoms. J Exp Bot 63:1543–1557
Qiu H, Yang EC, Bhattacharya D, Yoon HS (2012) Ancient gene paralogy may mislead inference of plastid phylogeny. Mol Biol Evol 29:3333–3343
Rice DW, Palmer JD (2006) An exceptional horizontal gene transfer in plastids: gene replacement by a distant bacterial paralog and evidence that haptophyte and cryptophyte plastids are sisters. BMC Biol 4:31
Ronquist F, Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19:1572–1574
Ruck EC, Nakov T, Jansen RK, Theriot EC, Alverson AJ (2014) Serial gene losses and foreign DNA underlie size and sequence variation in the plastid genomes of diatoms. Genome Biol Evol 6:644–654
Rutherford K, Parkhill J, Crook J, Horsnell T, Rice P, Rajandream MA, Barrell B (2000) Artemis: sequence visualization and annotation. Bioinformatics 16:944–945
Sagan L (1967) On the origin of mitosing cells. J Theor Biol 14:255–274
Sasaki NV, Sato N (2010) CyanoClust: comparative genome resources of cyanobacteria and plastids. Database 2010:bap025
Sato N (2000) SISEQ: manipulation of multiple sequence and large database files for common platforms. Bioinformatics 16:180–181
Sato N (2009) Gclust: trans-kingdom classification of proteins using automatic individual threshold setting. Bioinformatics 25:599–605
Silver MW, Mitchell JG, Ringo DL (1980) Siliceous nanoplankton. II. Newly discovered cysts and abundant choanoflagellates from the Weddell Sea, Antarctica. Mar Biol 58:211–217
Smith DR, Hua J, Lee RW, Keeling PJ (2012) Relative rates of evolution among the three genetic compartments of the red alga Porphyra differ from those of green plants and do not correlate with genome architecture. Mol Phylogenet Evol 65:339–344
Smith DR, Arrigo KR, Alderkamp AC, Allen AE (2014) Massive difference in synonymous substitution rates among mitochondrial, plastid, and nuclear genes of Phaeocystis algae. Mol Phylogenet Evol 71:36–40
Starkenburg SR, Kwon KJ, Jha RK, McKay C, Jacobs M, Chertkov O, Twary S, Rocap G, Cattolico RA (2014) A pangenomic analysis of the Nannochloropsis organellar genomes reveals novel genetic variations in key metabolic genes. BMC Genom 15:212
Tajima N, Sato S, Maruyama F, Kurokawa K, Ohta H, Tabata S, Sekine K, Moriyama T, Sato N (2014) Analysis of the complete plastid genome of the unicellular red alga Porphyridium purpureum. J Plant Res 127:389–397
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Whelan S, Goldman N (2001) A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol Biol Evol 18:691–699
Williams KP, Sobral BW, Dickerman AW (2007) A robust species tree for the alphaproteobacteria. J Bacteriol 189:4578–4586
Yang EC, Boo GH, Kim HJ, Cho SM, Boo SM, Andersen RA, Yoon HS (2012) Supermatrix data highlight the phylogenetic relationships of photosynthetic stramenopiles. Protist 163:217–231
Acknowledgments
This work was supported in part by a Grant-in-Aid for the Global COE Program, “From the Earth to Earths” and Scientific Research (No. 26291085) from the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan and the Canon Foundation (A. K. and N. S.).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. Kupiec.
This work is part of a dissertation submitted to the University of Tokyo in partial fulfillment of the requirements for the PhD degree.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Tajima, N., Saitoh, K., Sato, S. et al. Sequencing and analysis of the complete organellar genomes of Parmales, a closely related group to Bacillariophyta (diatoms). Curr Genet 62, 887–896 (2016). https://doi.org/10.1007/s00294-016-0598-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-016-0598-y