
Abstract
Destruxins are among the most exhaustively researched secondary metabolites of entomopathogenic fungi, yet definitive evidence for their roles in pathogenicity and virulence has yet to be shown. To establish the genetic bases for the biosynthesis of this family of depsipeptides, we identified a 23,792-bp gene in Metarhizium robertsii ARSEF 2575 containing six complete nonribosomal peptide synthetase modules, with an N-methyltransferase domain in each of the last two modules. This domain arrangement is consistent with the positioning of the adjacent amino acids N-methyl-l-valine and N-methyl-l-alanine within the depsipeptide structure of destruxin. DXS expression levels in vitro and in vivo exhibited comparable patterns, beginning at low levels during the early growth phases and increasing with time. Targeted gene knockout using Agrobacterium-mediated transformation produced mutants that failed to synthesize destruxins, in comparison with wild type and ectopic control strains, indicating the involvement of this gene in destruxin biosynthesis. The destruxin synthetase (DXS) disruption mutant was as virulent as the control strain when conidial inoculum was topically applied to larvae of Spodoptera exigua, Galleria mellonella, and Tenebrio molitor indicating that destruxins are dispensable for virulence in these insect hosts. The DXS mutants exhibited no other detectable changes in morphology and development.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Destruxins (DTX) are a family of cyclic depsipeptides produced by multiple plant and insect fungal pathogens (Pedras et al. 2002) (Fig. 1). The vast majority of the more than 35 members of the destruxin family have been isolated from strains belonging to the genus Metarhizium (Kodaira 1962; Pedras et al. 2002). These metabolites have been linked to a plethora of biological activities. Exogenously introduced DTX are highly toxic against a wide range of insect species (Amiri et al. 1999; Sree et al. 2008) and have been reported to accumulate in fungus-infected individuals (Suzuki et al. 1971; Vey and Goetz 1986). Injection, ingestion or topical application of purified DTX causes tetanic and flaccid paralysis, an effect linked to their abilities to depolarize Ca2+ gradients across the muscle plasma membrane (Samuels et al. 1988b; Dumas et al. 1996b; Hinaje et al. 2002), and the one that closely resembles symptoms observed in insects infected with destruxin-producing Metarhizium strains (Kodaira 1962; Samuels et al. 1988a). Intriguingly, DTX injected into insects also negatively impact both their cellular (Dumas et al. 1996b; Vilcinskas et al. 1997; Vey et al. 2002) and humoral (Pal et al. 2007) immune responses, which reinforces the notion of their potential contribution to Metarhizium virulence. These metabolites also induce oxidative stress (Sree and Padmaja 2008), damage midgut epithelium, disrupt salivary gland integrity (Sowjanya Sree and Padmaja 2008), and have a powerful inhibitory effect on fluid secretion by Malpighian tubules in various arthropods (James et al. 1993; Dumas et al. 1996b; Ruiz-Sanchez et al. 2010). Reports of different levels of DTX production in vitro by several Metarhizium strains and differential insect sensitivity to the metabolites have provided correlative data to explain the host range of those isolates, as well as their rapidity of killing (Samuels et al. 1988a; Kershaw et al. 1999; Moon et al. 2008; Wang et al. 2009). All of these results have contributed to the status of DTX as de facto virulence factors for those fungi that produce them. However, definitive genetic proof of their contributions to virulence has been lacking. In this paper, we describe the identification and inactivation by targeted gene knockout of a nonribosomal peptide synthetase (NRPS) gene from M. robertsii ARSEF 2575, demonstrate its role in DTX biosynthesis, and show that abolition of DTX production does not alter its virulence against larvae of the susceptible insect hosts Spodoptera exigua, Galleria mellonella, and Tenebrio molitor.

Chemical structure of destruxin A, B and E
Materials and methods
Strains, culture conditions, and Agrobacterium tumefaciens-mediated transformation (ATMT)
Metarhizium robertsii ARSEF 2575 (formerly known as M. anisopliae) (Bischoff et al. 2009) and its derivatives were grown on ¼-strength Sabouraud dextrose agar with yeast extract (SDAY/4) as previously described (Moon et al. 2008). For the analyses of destruxin production and in vitro gene expression, cultures were started with 1 × 106 conidia/100 ml HB broth (Giuliano Garisto Donzelli et al. 2010) and incubated in 250 ml flasks at 28°C in the dark on a rotary shaker at 150 rpm. ATMT and subsequent transformant handling were carried out as previously described, except that A. tumefaciens strain EHA105 was used instead of GV3101 (Moon et al. 2008; Giuliano Garisto Donzelli et al. 2010).
Cloning of the destruxin synthetase gene (DXS)
Based on the NCBI accession AJ273779 (Freimoser et al. 2003), we designed primers AJ273779F and AJ273779R (Table 1) and PCR screened a bacterial artificial chromosome (BAC) library constructed using M. robertsii ARSEF 2575 genomic DNA. BAC DNA from the positive clone 11F5 was partially sequenced at the Cornell University DNA Sequencing and Genotyping facility. The genomic region harboring the partial BAC sequence was identified using a draft of the M. robertsii ARSEF 2575 genome (Donzelli et al., unpublished).
Construction of pBDU-NRPS4KO
The binary vector pBDU-NRPS4KO was designed to target and disrupt the DXS gene. Two fragments from the DXS coding region (NRPS4-A and NRPS4-B, Fig. 5a), located 157 bp apart, were PCR amplified with the primer pairs NRPS4KO-A-F/NRPS4KO-A-R2 and NRPS4KO-B–F/NRPS4KO-B-R, respectively (Table 1), using ARSEF 2575 total DNA as the template. The bar expression cassette, which confers resistance to the drug bialaphos, was PCR amplified from pBARKS1 (Pall and Brunelli 1993) using the primers BarExprS-F and BarExprS-R-KS1 (Table 1). The binary vector pBDU-NRPS4KO was produced by assembling NRPS4-A, NRPS4-B, and bar into the XbaI- and Nt.BbvCI-linearized vector pBDU in the presence of the Uracil-Specific Excision Reagent or USER enzyme as suggested by the manufacturer (NEB, Ipswich, MA). pBDU is a pPK2 (Covert et al. 2001) derivative modified as suggested by NEB (http://www.neb.com/nebecomm/products/productN5504.asp.) to allow USER cloning. PCR amplifications for USER-based cloning were conducted as described (Nour-Eldin et al. 2006; Geu-Flores et al. 2007; Frandsen et al. 2008).
Screening for homologous recombination events at the DXS locus
Screening for homologous recombination events at the DXS locus was conducted by PCR and subsequently confirmed by Southern analysis. Screening primers for primary transformants were C6-ScreenF and Ptrpc80R (Table 1; Fig. 5a). PCR confirmation of both the DXS knockout (KO) event and purity of single conidial isolates made use of the primers NRPS4KO-screen-F and NRPS4KO-screen-R (Table 1; Fig. 5a). Southern analyses were carried out as previously described (Moon et al. 2008; Giuliano Garisto Donzelli et al. 2010) on SacI- or SalI-digested M. robertsii ARSEF 2575 total DNA. A PCR DIG Probe Synthesis Kit (Roche Applied Science, Indianapolis, IN), together with the primer couples NRPS4KO-screen-F/NRPS4KO-screen-R or BarF/BarR (Table 1), was used to synthesize DNA probes specific for either the NRPS4 (Probe 1, P1, Fig. 5a) or the bar (Probe 2, P2, Fig. 5a) coding regions, respectively.
RT-PCR for DXS expression
In vitro DXS expression was evaluated during growth in HB broth 19, 24, 48, 72, 96, 120, 168 and 240 h after medium inoculation with 1 ml of 106 conidia/ml harvested from 1-week-old cultures grown on SDAY/4. RNA extraction and reverse transcription conditions were as described (Giuliano Garisto Donzelli et al. 2010). Expression of DXS within fungus-infected S. exigua larvae at post-inoculation times of 28, 52, 72, 98, and 122 h, and 122-h non-motile larvae (early stage mummies, M1), 172-h non-motile larvae (late stage mummies, M2), and from controls 72 h after a mock inoculation followed a method previously described (Giuliano Garisto Donzelli et al. 2010). Primer couples used for the analyses were Baw-Act-275F/Baw-Act-439R, Ma-Btub F1/Ma-Btub-R18, and NRPS4KO-screen-F/NRPS4KO-screen-R, which anneal to the S. exigua β-tubulin, M. robertsii β-tubulin, and DXS genes, respectively (Table 1).
Detection of DTX
Production of DTX by the WT strain ARSEF 2575, the DXS KO strains 2.1 and 4.1, and the ectopic integrants 1.1 and 5.1 was determined by quantitative HPLC analysis of the major DTX components (A and B) in solid phase-extracted culture filtrate. All samples analyzed were from HB broth cultures grown for 5 days with rotary shaking (150 rpm) at 28°C. Solid phase extraction (SPE) was accomplished by loading 5 ml aliquots of culture filtrates onto a C18-SPE cartridge (200 mg; Alltech #40515) conditioned with 10 column volumes each of MeOH, and then deionized H2O. The charged cartridges were rinsed with 10 ml water and then eluted with 2 ml MeOH. The MeOH eluates were then dried under a nitrogen stream with gentle heating and reconstituted for analysis in 10 ml of MeOH for the positive controls (extracts from ARSEF 2575 and ectopic integrants), and 1 ml for the DTX-deficient extracts (DXS KO strains and uninoculated broth extracts). Thus, extracts of the KO strains and culture medium alone were analyzed at a tenfold higher concentration (relative to the culture filtrate volume) than those of the WT and ectopic strains, to insure detection of trace amounts of DTX, if present, in the former samples. The samples were analyzed by two different HPLC methods: (1) a relatively insensitive method using UV detection to estimate DTX production by the WT and ectopic strains, and (2) a more sensitive method using detection by MS–MS multiple reaction monitoring (MRM) to establish a lower limit of detection (LOD) for extracts of the KO strains.
In method 1, 10-μl aliquots of extract were injected onto an RP C18-A column (Varian Polaris, 250 mm × 4.6 mm, 5-μm particle, 180-Å pore), and eluted with acetonitrile:water (1:1) at a flow rate of 1 ml/min (Waters 600 pump), with detection by UV absorbance at 220 nm (extracted from a 190–350 nm scan on a Waters 996 diode array detector). DTX A and B were estimated using a standard curve for each compound. The LOD (established as S/N = 3) was 7- and 9-ng on-column, or 135- and 186-μg/L broth, for DTX A and B. DTX standards were purified from WT culture filtrates, and their identities were verified by 1H NMR on a Varian INOVA 600 spectrometer. DTX E, the third major component of the M. robertsii destruxin profile, has a labile epoxide group in the hydroxy acid side chain. It appears to co-vary quantitatively with DTX A and DTX B but, due to its instability, it was not included in the quantitative analysis.
In method 2, 5-μl aliquots were injected onto an RP-C18 column (Phenomenex Prodigy ODS3, 150 × 2 mm, 5-μm particle, 100-Å pore) eluted with acetonitrile:water:formic acid (650:350:1) at 0.25 ml/min, with detection by low resolution electrospray mass spectrometry on an ABI-Sciex Q-Trap 2000 spectrometer operated in positive ion mode. The pseudomolecular ions [M + H]+ of DTX A and B were observed at m/z 578 and 594, respectively. Accordingly, six MS–MS parent ion → product ion transitions were monitored, 578 → 465, 437, and 342 for detecting DTX A, and 594 → 481, 453, and 368 for detecting DTX B (as well as DTX E, which is isobaric to DTX B at unit mass resolution). Declustering and collision voltages used for all MS–MS experiments were 71 and 39 V, respectively. The LOD (S/N = 3) for this method was 12- and 16-pg on-column, or 0.5- and 0.6-ng/L broth for DTX A and B, respectively.
Insect virulence assays
Eggs of S. exigua were obtained from Benzon Research (Carlisle, PA). Newly hatched larvae were reared on BAW diet that contained chlortetracycline, methyl paraben and potassium sorbate (product no. F9220B, Bio-Serv, Frenchtown, NJ) at 25°C and 15:9 h light:dark regime until reaching the 2nd instar. Larvae were then transferred to Southland Beet Armyworm (BAW) diet containing chlortetracycline (Southland Products Inc., Lake Village, AR) as the only antibiotic until assay setup (Moon et al. 2008) and then moved to Southland BAW diet without chlortetracycline for the duration of the virulence assays.
Spodoptera exigua 2nd instar larvae were inoculated immediately after molting. G. mellonella and T. molitor larvae were purchased from Berkshire Biological (Westhampton, MA) and 4th instar individuals were inoculated within a few hours of arrival.
Conidia were harvested from 10-day-old cultures by addition of 10 ml of 0.1% Silwet L-77 (Loveland Industries Inc., Greeley, CO) and gently scraping the surface with a sterile inoculation loop to dislodge conidia. Conidial suspensions were pipetted from the plate and filtered through two layers of cheesecloth into sterile 50-ml tubes (Becton–Dickinson Falcon, Sparks, MD) and vortexed for 5 min. All larvae were dipped for 10 s in conidia resuspended in 0.1% Silwet L-77 at 104–107 conidia/ml, depending on the assay (Table 3). Larvae dipped in 0.1% Silwet-L-77 alone were used as controls. In all instances, 24 larvae/treatment were assayed. Mortality and time-to-death inflicted by the WT and the DXS KO strain 4.1 were evaluated up to 7 days after inoculation. Two or three independent assays were carried out for S. exigua (two), G. mellonella (three) and T. molitor (three). After dipping, larvae were placed individually in 24-well plates with diet and incubated at 25°C with a 15:9 h light:dark photoperiod. Larvae were subsequently transferred to clean plates and provided fresh diet every day to avoid uncontrolled re-inoculation from fungus growing on diet, frass and exuviae.
Data analysis
Identification of functional domains in DXS and surrounding genes was carried out using InterProScan (Quevillon et al. 2005) and PFAM (Finn et al. 2010). N-methyltransferase domains were identified using motifs previously described (Weber et al. 1994; Ansari et al. 2008) and confirmed by comparing N-methyltransferase domains extracted from NRPSs with known N-methylated products to C- and O-methyltransferase domains from other proteins. To this end, multiple sequence alignment was carried out with Muscle (Edgar 2004), followed by cluster analysis based on the Maximum Likelihood method and WAG model, which assumed a Gamma distribution calculated on five discrete categories and a shape parameter of 4.3882, calculated using 180 positions in the available dataset and 500 bootstrap replicates. Both steps were conducted with the appropriate modules provided in MEGA5 (Tamura et al. 2011).
Data from insect virulence assays were analyzed using the survival analysis module provided with JMP 9.0.2 (SAS Institute Inc. Cary, NC). Survival curves calculated for insects inoculated with either the DXS KO strain 4.1 or the WT were compared with the Log-rank test.
Results
DXS gene isolation and structure
Initial cloning of the DXS gene was carried out using the cDNA sequence of NCBI accession number AJ273779. This clone was previously identified (Freimoser et al. 2003) from M. anisopliae ARSEF 2575 grown for 24 h in minimal medium containing 1% cockroach cuticles. Partial sequencing of a BAC clone containing AJ273779 yielded a ~14,000-bp sequence, which included the 3′ end of an uncharacterized NRPS containing two N-methyltransferase domains. Complete gene structure, determined using data from an early draft of the M. robertsii ARSEF 2575 genome (Donzelli et al., unpublished data), revealed a 23,792-nt ORF, likely interrupted by one intron, which encoded a predicted 7,913 aa polypeptide (GenBank accession JN805540). The gene (DXS) was located in a 107,640-nt contig of the ARSEF 2575 genome in which a second, 2-module GliP-like NRPS and a polyketide synthase (PKS) predicted to synthesize a reduced polyketide were also present (Fig. 2a). BlastN similarity searches indicated that the region is nearly perfectly conserved in M. anisopliae ARSEF 23 (scaffold 50), while no significant matches were detected in the M. acridum CQMa 102 genome (Gao et al. 2011).

a Structure and predicted specificity of each module of DXS. A adenylation domain, T thiolation domain, C condensation domain, M N-methyltransferase domain. b Gene models identified in the genomic region surrounding DXS. For each predicted gene, the putative protein product, its predicted amino acid length, and the best non-Metarhizium BlastX match in the nr database are reported
DXS contains 6 complete NRPS modules, with an N-methyltransferase domain in each of the last two modules (Fig. 2b). Both domains were aligned with corresponding amino acid sequences retrieved from several fungal NRPSs producing N-methylated products and with C-methyltransferase domains from several hybrid PKS–NRPSs. Similar to what has been described by others (Ansari et al. 2008), both putative N-methyltransferase domains from DXS clustered with their homologs in other organisms and displayed motifs identified previously (Weber et al. 1994; Ansari et al. 2008) (Fig. 3a). The position of the N-methyltransferase domains and the directionality of peptide synthesis were used to infer the specificity of each adenylation domain (Figs. 2b, 3b). The assignment is further supported by the sequence divergence of the putative hydroxy acid (hac) adenylation domain (A2) from other typical amino acid-activating A domains (Fig. 3b).

a Cluster analysis of N-methyltransferase (NMet) domains extracted from NRPSs having known N-methylated products, which included destruxin, DXS, cyclosporine, CssA (Weber et al. 1994), aureobasidin A, AbA (Slightom et al. 2009), enniatin, ESYN (Haese et al. 1993), beauvericin, BEAS (Xu et al. 2008), and bassianolide, BasSY (Jirakkakul et al. 2008); C-methyltransferase domains extracted from several PKSs encoding C-methylated products (Skellam et al. 2010), including compactin, mlcA and mlcB (Eisfeld 2009), lovastatin, LNKS (Hendrickson et al. 1999) and LDKS (Kennedy et al. 1999), fusarin C, FUSS (Song et al. 2004), cytochalasan, CheA (Schumann and Hertweck 2007); and the O-methyltransferases involved in the biosynthesis of gliotoxin, GliM (Gardiner and Howlett 2005), aflatoxin, stcP (Kelkar et al. 1996), and apicidin, APS6 (Jin et al. 2010). Note that the four NMet domains in AbA are 100% identical to each other at the amino acid level, so only one was used for this analysis. The tree was inferred using the Maximum Likelihood method based on the WAG model and 500 bootstrap replicates. The bootstrap value (percentage of replicate trees showing that same clade) is reported at each node of the tree. The GenBank accession number and the protein segment used for the analysis are reported at the end of each branch. b Comparison of the 10-aa code (Stachelhaus et al. 1999) of the six DXS adenylation domains with those from other fungal synthetases having known metabolites. CssA: cyclosporine synthetase from Tolypocladium inflatum (CAA82227), AbA: aurobasidin A from Aureobasidium pullulans (ACJ04424), feESYN: enniatin synthetase from Fusarium equiseti (CAA79245.2), fsESYN: enniatin synthetase from Fusarium sambucineum (Pieper et al. 1992; Xu et al. 2009), and Ba1: bacitracin synthetase 1 from Bacillus licheniformis (O68006). The 10-aa code was identified using NRPSpredictor (Rausch et al. 2005)
In vitro and in vivo expression of DXS
Expression of DXS was characterized by RT-PCR both in vitro and during the interaction with the S. exigua host. In both cases, the transcription pattern was relatively simple: DXS expression was low during early growth phases (i.e., after the first 48 h in HB broth and 72 h into S. exigua infection), increased with time under both conditions, and reached a steady high level at later stages (Fig. 4a, b). RT-PCR also indicated the presence of DXS transcripts in conidia (Fig. 4a, time 0).

Detection of DXS expression by RT-PCR in Metarhizium robertsii a conidia (time 0) or mycelia 19, 24, 48, 72, 96, 120, 168 and 240 h after inoculation into HB medium; and b in fungus-infected motile Spodoptera exigua larvae 28, 52, 72, 98, and 122 h after inoculation, and from fungus-infected non-motile larvae 122 h (M1) and 172 h (M2) after inoculation. RNA extracted from mock-inoculated larvae at 72 h was used as a control (C)
Identification of DXS knockout strains
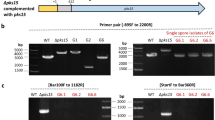
Initial PCR-screening of bialaphos-resistant colonies and subsequent confirmation of DXS disruption after selection of single conidial progeny (Fig. 5a, b) lead to the identification of strains 2.1 and 4.1 as homologous integrants at the DXS locus. The bialaphos-resistant strains 1.1 and 5.1, in which DXS appeared to be intact, were retained as phenotypic controls. Southern analysis confirmed the PCR analysis demonstrating that 2.1 and 4.1 carried a single T-DNA insertion at the DXS locus (Fig. 5c). Southern analysis also confirmed the integration of the T-DNA outside the targeted region in strains 1.1 and 5.1 (Fig. 5c).

a Recombination of the bar gene within the DXS gene to create DXS knockout (KO) strains. Position of the primers used for PCR identification and confirmation of the DXS KO events is marked by small black arrows. Position of the probes used for Southern analyses is also indicated and marked as P1 (DXS probe) or P2 (bar probe). The predicted amino acid specificity of the depicted adenylation domains is reported. b DXS disruption detected by PCR in strains 2.1 and 4.1 using the primers NRPS4KO-screen-F and NRPS4KO-screen-R (Table 1). Wild type (WT) strain ARSEF 2575 and ectopic integrants 1.1 and 5.1 were used as the negative controls. T-DNA insertion within the DXS coding region is revealed by the increased size to 2,242 bp due to the insertion of the 1,867-bp bar expression cassette into the 2.1 and 4.1 amplicons compared to the 532-bp amplification product obtained from the wild type (WT) and ectopic integrants. c Southern analyses comparing WT, the ectopic integrant strains 1.1 and 5.1, and the homologous integrant strains 2.1 and 4.1. Genomic DNA of each strain (2 μg) was digested with either SalI or SacI and hybridized with either the DXS or bar probe after gel separation and transfer onto nylon membrane
Phenotypic effects of the DXS knockout
DXS KO strains 2.1 and 4.1 were analyzed for production of DTX and both failed to yield detectable amounts of the metabolites (Fig. 6; Table 2). The ectopic integrants 5.1 and 1.1, on the other hand, retained the ability to synthesize these metabolites at WT levels (Fig. 6; Table 2). No other obvious phenotypic changes were observed in the mutants, including pigmentation, growth rate, or morphology (data not shown). Abolition of DTX biosynthesis had no significant effect on virulence levels of M. robertsii ARSEF 2575 against larvae of S. exigua (2nd instar), G. mellonella (4th instar) and T. molitor (4th instar) at any of the wide range of inoculum concentrations tested and in independently replicated assays with one exception. In assay #1 with T. molitor, the KO strain was significantly less virulent than the WT when the lowest inoculum dose was applied (Table 3), but this result was not repeated in the following two assays (Table 3).

HPLC-MS–MS analyses of culture filtrate extracts from the wild type (WT) ARSEF 2575 (2.5 μl broth equivalent), the ectopic integrants 1.1 and 5.1 (2.5 μl broth equivalent), and the DXS null mutants 2.1 and 4.1 (25 μl broth equivalent), grown in HB broth for 5 days; an extract of the uninoculated broth was included as a negative control (25 μl broth equivalent). Note that the KO strain samples and the broth control were injected at a tenfold higher concentration relative to the extracted broth volume than that of the WT and ectopic samples. Also included for comparison is a standard composed of pure destruxins A and B (500 pg each loaded onto the column). Chromatograms are summed ion traces from 6 MS–MS transitions (594 → 481, 453, 368 for detecting DTX E and B; 578 → 465, 437, 342 for detecting DTX A). Peak intensity is measured in ion counts per second (cps). The DTX A, B and E peaks are marked with the corresponding letters
Discussion
Destruxins A, B and E are the primary constituents reported from fermentation broths of M. robertsii, and they are by far the most exhaustively researched toxins of entomopathogenic fungi. Using targeted gene knockout, we identified DXS as the primary gene responsible for DTX biosynthesis. DXS encodes for an NRPS, a family of large multifunctional, multimodular enzymes. Each NRPS module contains domains needed for the activation (adenylation domain), propagation (thiolation domain), and incorporation (condensation domain) of one monomer contained in the final metabolite. In some cases, the process can be iterative and one module introduces an amino acid multiple times in the final molecule but, in the most frequent cases to date, fungal NPRSs assemble the peptide in a linear fashion, adding one amino acid for each module in the protein (Eisfeld 2009). Consistent with DTX structure, DXS encodes for a large protein carrying six NRPS modules, where each module accounts for the incorporation of one of the six monomers (five amino acids and one hydroxy acid) constituting the DTX backbone structure (Fig. 1). The presence of N-methyltransferase domains in the last two DXS modules fits well with the presence of N-methyl-l-valine and N-methyl-l-alanine in the DTX molecule (Fig. 1), while their locations allow the putative attribution of the amino acid specificity of each DXS module, as shown in Fig. 2b. Heterogeneity in the DTX family of cyclic depsipeptides is conferred by variation in the hydroxy acid and the amino acid substitutions in positions 1–4. We anticipate that fungi capable of producing structural variants of DTX not found in M. robertsii ARSEF 2575 will likely have slightly modified modules that account for the reported amino acid substitutions (Pedras et al. 2002).
The role of DXS in DTX biosynthesis is supported by the absence of the metabolites in independently disrupted mutants as compared to the WT and ectopic strains. In addition, the genome of M. acridum, known to be a non-producer of DTX (Kershaw et al. 1999; Moon et al. 2008), does not contain any homologs of this gene and its surrounding region (Gao et al. 2011).
The similarity of the in vivo and in vitro expression patterns of DXS, as observed in Fig. 4, suggests a developmental regulation of this gene, similar to what has been reported with other secondary metabolites (Calvo et al. 2002; Kato et al. 2003; Yu and Keller 2005; Fox and Howlett 2008) including those from M. robertsii (Giuliano Garisto Donzelli et al. 2010). DXS transcripts were also detected in conidia produced on SDAY/4, hinting at either the presence of the metabolites in these propagules or a possible accumulation of transcripts during conidium formation.
Loss of DTX production had no phenotypic effect in our experimental settings. This included no stable changes in virulence against any of the three insect species assayed, which is surprising considering the existing body of work supporting the insecticidal effects of DTXs. Others have drawn a tentative link between the ability of some Metarhizium strains to rapidly kill their hosts (Samuels et al. 1988a; Amiri-Besheli et al. 2000) and their production of secondary metabolites, including DTX. However, the relationship between fungal secondary metabolism and pathogenicity is far from understood and, for the vast majority of cases, the roles of small molecules in these complex processes remain elusive. For instance, hybrid PKS–NRPS pathways leading to the biosynthesis of tenellin in Beauveria bassiana and the mutagenic fusarin-like NG-39X compounds in M. robertsii were found not to contribute significantly to virulence against G. mellonella and S. exigua larvae, respectively (Eley et al. 2007; Giuliano Garisto Donzelli et al. 2010). Similarly, targeted gene disruption experiments with M. robertsii ARSEF 2575 to test the role of serinocyclin, a spore-derived peptide, revealed that KO strains were as virulent as WT when assayed on larvae of S. exigua or Leptinotarsa decemlineata, and no differences in morphology or physiology could be demonstrated (Moon et al. 2008). In contrast, mutants of B. bassiana with targeted disruptions of the peptide synthetase genes responsible for the biosynthesis of beauvericin and bassianolide were morphologically indistinguishable from WT, but the KO strains showed decreased virulence when tested against S. frugiperda, Helicoverpa zeae, and G. mellonella larvae (Xu et al. 2008, 2009). Similarly, targeted gene knockout of a geranylgeranyl diphosphate synthase abolished the production of helvolic acid and reduced virulence of M. anisopliae NAFF635007 against two genera of insect larvae (Singkaravanit et al. 2010).
The finding that the genetic abolishment of DTX production in M. robertsii had no measurable effect on the virulence of the fungus when conidia were applied topically to S. exigua, G. mellonella or T. molitor larvae is in stark contrast to the outcome predicted by a large body of in vitro toxicological studies suggesting DTX are key virulence factors for Metarhizium invertebrate pathogens (Pal et al. 2007; Sree and Padmaja 2008). Resolving this incongruity will require further investigation. For instance, some insects can recover from sub-lethal DTX applications, and sensitivity to DTX has been shown to vary with the host species (Samuels et al. 1988a; Kershaw et al. 1999). However, little is known about possible detoxification mechanisms of most arthropods and whether they are constitutive or induced as part of the immune response mounted by the insect after fungal infection (Rohlfs and Churchill 2011). It is also possible that DTXs are contributory virulence factors that affect only hosts carrying specific-molecular targets, as seen for some secondary metabolites produced by plant pathogenic fungi (Sindhu et al. 2008; Sweat et al. 2008), or their presence may be redundant for virulence because of the action of additional unknown compounds produced in amounts sufficient to ensure efficient killing, the effects of which mask a specific role for DTX. This is supported by the occasional inconsistent correlation between strain virulence and in vitro DTX production (Samuels et al. 1988a; Kershaw et al. 1999; Amiri-Besheli et al. 2000; Moon et al. 2008).
It has been reported that loss of a dispensable chromosome in M. anisopliae strain V275 resulted in several biochemical changes including the inability to produce DTX; the V275 strain showed reduced virulence on T. molitor, but there were no differences against G. mellonella. However, the number of phenotypic changes associated with dispensable chromosome loss does not allow one to attribute the observed difference in virulence solely to the loss of DTX production (Wang et al. 2003). Our data suggest that the virulence changes observed in strain V275 were likely unrelated to the abolishment of DTX production, since we report here no loss in virulence of a DTX-deficient KO strain against T. molitor larvae. In addition, there is an abundance of genes encoding potentially toxic proteins and putative secondary metabolites predicted from the recently sequenced M. anisopliae ARSEF 23 and M. robertsii ARSEF 2575 genomes, many of which might be new candidates for playing causal roles in virulence (Gao et al. 2011; Donzelli et al., unpublished). Alternatively, the relationship of DTX to virulence may be coincidental, and the compounds may play roles in other unknown functions, or cause subtle effects in the host, not measurable with traditional pathogenicity assays. Our study neither confirms nor refutes these hypotheses definitively but clearly demonstrates the dispensability of DTX as virulence factors of M. robertsii ARSEF 2575 as a pathogen of S. exigua, T. molitor, and G. mellonella.
References
Amiri B, Ibrahim L, Butt TM (1999) Antifeedant properties of destruxins and their potential use with the entomogenous fungus Metarhizium anisopliae for improved control of crucifer pests. Biocontrol Sci Technol 9:487–498
Amiri-Besheli B, Khambay B, Cameron S, Deadman ML, Butt TM (2000) Inter- and intra-specific variation in destruxin production by insect pathogenic Metarhizium spp., and its significance to pathogenesis. Mycol Res 104:447–452
Ansari MZ, Sharma J, Gokhale RS, Mohanty D (2008) In silico analysis of methyltransferase domains involved in biosynthesis of secondary metabolites. BMC Bioinform 9:454
Bischoff JF, Rehner SA, Humber RA (2009) A multilocus phylogeny of the Metarhizium anisopliae lineage. Mycologia 101:512–530
Calvo AM, Wilson RA, Bok JW, Keller NP (2002) Relationship between secondary metabolism and fungal development. Microbiol Mol Biol Rev 66:447–459
Covert SF, Kapoor P, Lee MH, Briley A, Nairn CJ (2001) Agrobacterium tumefaciens-mediated transformation of Fusarium circinatum. Mycol Res 105:259–264
Dumas C, Matha V, Quiot JM, Vey A (1996a) Effects of destruxins, cyclic depsipeptide mycotoxins, on calcium balance and phosphorylation of intracellular proteins in lepidopteran cell lines. Comp Biochem Physiol C Comp Pharmacol Toxicol 114:213–219
Dumas C, Ravallec M, Matha V, Vey A (1996b) Comparative study of the cytological aspects of the mode of action of destruxins and other peptidic fungal metabolites on target epithelial cells. J Invertebr Pathol 67:137–146
Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797
Eisfeld K (2009) Non-ribosomal peptide synthetases of fungi. In: Anke T, Weber D (eds) The mycota, physiology and genetics. Springer, Heidelberg, pp 305–330
Eley KL, Halo LM, Song Z, Powles H, Cox RJ, Bailey AM, Lazarus CM, Simpson TJ (2007) Biosynthesis of the 2-pyridone tenellin in the insect pathogenic fungus Beauveria bassiana. ChemBioChem 8:289–297
Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, Holm L, Sonnhammer EL, Eddy SR, Bateman A (2010) The Pfam protein families database. Nucleic Acids Res 38:D211–D222
Fox EM, Howlett BJ (2008) Secondary metabolism: regulation and role in fungal biology. Curr Opin Microbiol 11:481–487
Frandsen RJ, Andersson JA, Kristensen MB, Giese H (2008) Efficient four fragment cloning for the construction of vectors for targeted gene replacement in filamentous fungi. BMC Mol Biol 9:70
Freimoser FM, Screen S, Bagga S, Hu G, St. Leger RJ (2003) Expressed sequence tag (EST) analysis of two subspecies of Metarhizium anisopliae reveals a plethora of secreted proteins with potential activity in insect hosts. Microbiology 149:239–247
Gao Q, Jin K, Ying SH, Zhang Y, Xiao G, Shang Y, Duan Z, Hu X, Xie XQ, Zhou G, Peng G, Luo Z, Huang W, Wang B, Fang W, Wang S, Zhong Y, Ma LJ, St. Leger RJ, Zhao GP, Pei Y, Feng MG, Xia Y, Wang C (2011) Genome sequencing and comparative transcriptomics of the model entomopathogenic fungi Metarhizium anisopliae and M. acridum. PLoS Genet 7:e1001264
Gardiner DM, Howlett BJ (2005) Bioinformatic and expression analysis of the putative gliotoxin biosynthetic gene cluster of Aspergillus fumigatus. FEMS Microbiol Lett 248:241–248
Geu-Flores F, Nour-Eldin HH, Nielsen MT, Halkier BA (2007) USER fusion: a rapid and efficient method for simultaneous fusion and cloning of multiple PCR products. Nucleic Acids Res 35:e55
Giuliano Garisto Donzelli B, Krasnoff SB, Churchill ACL, Vandenberg JD, Gibson DM (2010) Identification of a hybrid PKS-NRPS required for the biosynthesis of NG-391 in Metarhizium robertsii. Curr Genet 56:151–162
Haese A, Schubert M, Herrmann M, Zocher R (1993) Molecular characterization of the enniatin synthetase gene encoding a multifunctional enzyme catalysing N-methyldepsipeptide formation in Fusarium scirpi. Mol Microbiol 7:905–914
Hendrickson L, Davis CR, Roach C, Nguyen DK, Aldrich T, McAda PC, Reeves CD (1999) Lovastatin biosynthesis in Aspergillus terreus: characterization of blocked mutants, enzyme activities and a multifunctional polyketide synthase gene. Chem Biol 6:429–439
Hinaje M, Ford M, Banting L, Arkle S, Khambay B (2002) An investigation of the ionophoric characteristics of destruxin A. Arch Biochem Biophys 405:73–77
James PJ, Kershaw MJ, Reynolds SE, Charnley AK (1993) Inhibition of desert locust (Schistocerca gregaria) Malpighian tubule fluid secretion by destruxins, cyclic peptide toxins from the insect pathogenic fungus Metarhizium anisopliae. J Insect Physiol 39:797–804
Jin JM, Lee S, Lee J, Baek SR, Kim JC, Yun SH, Park SY, Kang S, Lee YW (2010) Functional characterization and manipulation of the apicidin biosynthetic pathway in Fusarium semitectum. Mol Microbiol 76:456–466
Jirakkakul J, Punya J, Pongpattanakitshote S, Paungmoung P, Vorapreeda N, Tachaleat A, Klomnara C, Tanticharoen M, Cheevadhanarak S (2008) Identification of the nonribosomal peptide synthetase gene responsible for bassianolide synthesis in wood-decaying fungus Xylaria sp. BCC1067. Microbiology 154:995–1006
Kato N, Brooks W, Calvo AM (2003) The expression of sterigmatocystin and penicillin genes in Aspergillus nidulans is controlled by veA, a gene required for sexual development. Eukaryot Cell 2:1178–1186
Kelkar HS, Keller NP, Adams TH (1996) Aspergillus nidulans stcP encodes an O-methyltransferase that is required for sterigmatocystin biosynthesis. Appl Environ Microbiol 62:4296–4298
Kennedy J, Auclair K, Kendrew SG, Park C, Vederas JC, Hutchinson CR (1999) Modulation of polyketide synthase activity by accessory proteins during lovastatin biosynthesis. Science 284:1368–1372
Kershaw MJ, Moorhouse ER, Bateman R, Reynolds SE, Charnley AK (1999) The role of destruxins in the pathogenicity of Metarhizium anisopliae for three species of insect. J Invertebr Pathol 74:213–223
Kodaira Y (1962) Studies on the new toxic substances to insects, destruxin A and B, produced by Oospora destructor. Agric Biol Chem 26:39–62
Moon YS, Donzelli BG, Krasnoff S, McLane H, Griggs MH, Cooke P, Vandenberg JD, Gibson DM, Churchill ACL (2008) Agrobacterium-mediated disruption of a nonribosomal peptide synthetase gene in the invertebrate pathogen Metarhizium anisopliae reveals a peptide spore factor. Appl Environ Microbiol 74:4366–4380
Nour-Eldin HH, Hansen BG, Norholm MH, Jensen JK, Halkier BA (2006) Advancing uracil-excision based cloning towards an ideal technique for cloning PCR fragments. Nucleic Acids Res 34:e122
Pal S, St. Leger RJ, Wu LP (2007) Fungal peptide destruxin A plays a specific role in suppressing the innate immune response in Drosophila melanogaster. J Biol Chem 282:8969–8977
Pall ML, Brunelli JP (1993) A series of six compact fungal transformation vectors containing polylinkers with multiple unique restriction sites. Fungal Genet News 40:59–62
Pedras MSC, Zaharia IL, Ward DE (2002) The destruxins: synthesis, biosynthesis, biotransformation, and biological activity. Phytochemistry 59:579–596
Pieper R, Kleinkauf H, Zocher R (1992) Enniatin synthetases from different fusaria exhibiting distinct amino acid specificities. J Antibiot (Tokyo) 45:1273–1277
Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, Apweiler R, Lopez R (2005) InterProScan: protein domains identifier. Nucleic Acids Res 33:W116–W120
Rausch C, Weber T, Kohlbacher O, Wohlleben W, Huson DH (2005) Specificity prediction of adenylation domains in nonribosomal peptide synthetases (NRPS) using transductive support vector machines (TSVMs). Nucleic Acids Res 33:5799–5808
Rohlfs M, Churchill ACL (2011) Fungal secondary metabolites as modulators of interactions with insects and other arthropods. Fungal Genet Biol 48:23–34
Ruiz-Sanchez E, Orchard I, Lange AB (2010) Effects of the cyclopeptide mycotoxin destruxin A on the Malpighian tubules of Rhodnius prolixus (Stål). Toxicon 55:1162–1170
Samuels RI, Charnley AK, Reynolds SE (1988a) The role of destruxins in the pathogenicity of 3 strains of Metarhizium anisopliae for the tobacco hornworm Manduca sexta. Mycopathologia 104:51–58
Samuels RI, Reynolds SE, Charnley AK (1988b) Calcium channel activation of insect muscle by destruxins, insecticidal compounds produced by the entomopathogenic fungus Metarhizium anisopliae. Comp Biochem Physiol C Comp Pharmacol Toxicol 90:403–412
Schumann J, Hertweck C (2007) Molecular basis of cytochalasan biosynthesis in fungi: gene cluster analysis and evidence for the involvement of a PKS-NRPS hybrid synthase by RNA silencing. J Am Chem Soc 129:9564–9565
Sindhu A, Chintamanani S, Brandt AS, Zanis M, Scofield SR, Johal GS (2008) A guardian of grasses: specific origin and conservation of a unique disease-resistance gene in the grass lineage. Proc Natl Acad Sci USA 105:1762–1767
Singkaravanit S, Kinoshita H, Ihara F, Nihira T (2010) Cloning and functional analysis of the second geranylgeranyl diphosphate synthase gene influencing helvolic acid biosynthesis in Metarhizium anisopliae. Appl Microbiol Biotechnol 87:1077–1088
Skellam EJ, Hurley D, Davison J, Lazarus CM, Simpson TJ, Cox RJ (2010) Mutation of key residues in the C-methyltransferase domain of a fungal highly reducing polyketide synthase. Mol Biosyst 6:680–682
Slightom JL, Metzger BP, Luu HT, Elhammer AP (2009) Cloning and molecular characterization of the gene encoding the aureobasidin A biosynthesis complex in Aureobasidium pullulans BP-1938. Gene 431:67–79
Song Z, Cox RJ, Lazarus CM, Simpson TT (2004) Fusarin C biosynthesis in Fusarium moniliforme and Fusarium venenatum. ChemBioChem 5:1196–1203
Sowjanya Sree K, Padmaja V (2008) Oxidative stress induced by destruxin from Metarhizium anisopliae (Metch.) involves changes in glutathione and ascorbate metabolism and instigates ultrastructural changes in the salivary glands of Spodoptera litura (Fab.) larvae. Toxicon 51:1140–1150
Sree KS, Padmaja V (2008) Destruxin from Metarhizium anisopliae induces oxidative stress effecting larval mortality of the polyphagous pest Spodoptera litura. J Appl Entomol 132:68–78
Sree KS, Padmaja V, Murthy YLN (2008) Insecticidal activity of destruxin, a mycotoxin from Metarhizium anisopliae (Hypocreales), against Spodoptera litura (Lepidoptera: Noctuidae) larval stages. Pest Manag Sci 64:119–125
Stachelhaus T, Mootz HD, Marahiel MA (1999) The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem Biol 6:493–505
Suzuki A, Kawakami K, Tamura S (1971) Detection of destruxins in silkworm larvae infected with Metarrhizium anisopliae. Agric Biol Chem 35:1641–1643
Sweat TA, Lorang JM, Bakker EG, Wolpert TJ (2008) Characterization of natural and induced variation in the LOV1 gene, a CC-NB-LRR gene conferring victorin sensitivity and disease susceptibility in Arabidopsis. Mol Plant Microbe Interact 21:7–19
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Vey A, Goetz P (1986) Antifungal cellular defense mechanisms in insects. In: Gupta AP (ed) Hemocytic and Humoral Immunity in Arthropods. Wiley, New York, pp 89–115
Vey A, Matha V, Dumas C (2002) Effects of the peptide mycotoxin destruxin E on insect haemocytes and on dynamics and efficiency of the multicellular immune reaction. J Invertebr Pathol 80:177–187
Vilcinskas A, Matha V, Götz P (1997) Inhibition of phagocytic activity of plasmatocytes isolated from Galleria mellonella by entomogenous fungi and their secondary metabolites. J Insect Physiol 43:475–483
Wang C, Skrobek A, Butt TM (2003) Concurrence of losing a chromosome and the ability to produce destruxins in a mutant of Metarhizium anisopliae. FEMS Microbiol Lett 226:373–378
Wang S, Leclerque A, Pava-Ripoll M, Fang W, St. Leger RJ (2009) Comparative genomics using microarrays reveals divergence and loss of virulence-associated genes in host-specific strains of the insect pathogen Metarhizium anisopliae. Eukaryot Cell 8:888–898
Weber G, Schorgendorfer K, Schneider-Scherzer E, Leitner E (1994) The peptide synthetase catalyzing cyclosporine production in Tolypocladium niveum is encoded by a giant 45.8-kilobase open reading frame. Curr Genet 26:120–125
Xu Y, Orozco R, Wijeratne EM, Gunatilaka AA, Stock SP, Molnar I (2008) Biosynthesis of the cyclooligomer depsipeptide beauvericin, a virulence factor of the entomopathogenic fungus Beauveria bassiana. Chem Biol 15:898–907
Xu Y, Orozco R, Wijeratne KEM, Espinosa-Artiles P, Gunatilaka LAA, Stock PS, Molnar I (2009) Biosynthesis of the cyclooligomer depsipeptide bassianolide, an insecticidal virulence factor of Beauveria bassiana. Fungal Genet Biol 46:353–364
Yu JH, Keller N (2005) Regulation of secondary metabolism in filamentous fungi. Annu Rev Phytopathol 43:437–458
Acknowledgments
We thank Andy M. Bailey (University of Bristol, UK) for generously providing the sequences of several unpublished NRPS fragments from M. robertsii, one of which was used for closing a sequencing gap during the initial cloning of the DXS gene. This project was supported in part by the National Research Initiative of the USDA Cooperative State Research, Education and Extension Service, grant numbers 2002-35316-12207 (awarded to D.M. Gibson, A.C.L. Churchill, and J.D. Vandenberg) and 2005-35607-15283 (awarded to A.C.L. Churchill, D.M. Gibson, and J.D. Vandenberg), and by the USDA Postdoctoral Associate Program (awarded to D.M. Gibson). While this manuscript was under review we became aware that the article entitled “Unveiling the biosynthetic puzzle of destruxins in Metarhizium species” by Bing Wang, Qianjin Kanf, Linguan Bai, and Chengshu Wang had been published in PNAS vol. 109 no. 4. The findings described in this paper and in our work are in part overlapping. An early summary of this work was presented at the 26th Fungal Genetics Conference at Asilomar March 15–20, 2011, poster #607.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by A. Brakhage.
An erratum to this article can be found at http://dx.doi.org/10.1007/s00294-012-0371-9.
Rights and permissions
About this article
Cite this article
Giuliano Garisto Donzelli, B., Krasnoff, S.B., Sun-Moon, Y. et al. Genetic basis of destruxin production in the entomopathogen Metarhizium robertsii . Curr Genet 58, 105–116 (2012). https://doi.org/10.1007/s00294-012-0368-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-012-0368-4