
Abstract
In this study, we conducted a meta-analysis on 16S rRNA gene sequences of bovine fecal origin that are publicly available in the RDP database. A total of 13,663 sequences including 603 isolate sequences were identified in the RDP database (Release 11, Update 1), where 13,447 sequences were assigned to 10 phyla, 17 classes, 28 orders, 59 families, and 110 genera, while the remaining 216 sequences could not be assigned to a known phylum. Firmicutes and Bacteroidetes were the first and the second predominant phyla, respectively. About 41 % of the total sequences could not be assigned to a known genus. The total sequences were assigned to 1252 OTUs at 97 % sequence similarity. A small number of OTUs shared among datasets indicate that fecal bacterial communities of cattle are greatly affected by various factors, specifically diet. This study may guide future studies to further analyze fecal bacterial communities of cattle.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Fecal bacterial communities of cattle have an influence on animal health and food safety. Diet was the greatest factor altering fecal bacterial communities, while breed, gender, age, and macroecological factors were minor factors altering fecal bacterial communities [8]. Callaway et al. [1] investigated fecal bacterial communities from 6 cattle fed 0, 25, or 50 % dried distillers’ grain (2 cattle per diet), while Shanks et al. [19] investigated fecal bacterial communities recovered from 30 cattle that were equally divided into six cattle populations (3 diet groups × 2 locations; 5 animals per population analyzed). Rice et al. [17] used the next-generation pyrosequencing method to examine fecal bacterial communities recovered from 20 cattle fed 5 diets with differing types and levels of distillers’ grains, and the structure of the fecal microbiota of the distillers’ grain-based diets observed was significantly different from that of the control diet. These three studies showed that fecal bacterial communities were affected by diet. In addition, there was variation among individual animals although the same diet was fed to the cattle [3, 4]. Therefore, better understanding of bovine fecal bacteria affected by these factors would be important in animal health and food safety.
Although a recent study isolated some fecal bacteria of cattle [27], few culture-dependent studies to date have been conducted to isolate and characterize bovine fecal bacteria. After 16S rRNA gene sequences were used as a culture-independent approach [26], fecal bacterial communities have been analyzed using cloning and the Sanger sequencing technology [4, 14]. However, individual studies using the Sanger sequencing technology to date focused on a specific fecal bacterial ecosystem and showed only a small portion of the whole diversity present in cattle feces. In addition, taxa of 16S rRNA gene sequences recovered in the individual studies were analyzed using old versions of taxonomy classifier with less robust sequence libraries. Therefore, individual studies likely bias the knowledge on fecal bacterial communities. In the current study, we conducted a meta-analysis to provide a collective view of the fecal bacterial communities using the collective 16S rRNA gene sequences that are publicly available in the RDP database. We also predicted the current coverage of fecal bacterial diversity of cattle based on the rarefaction estimate.
Materials and Methods
Collection of Sequence Datasets
All the 16S rRNA gene sequences recovered from cattle feces were collected from the publicly available RDP database (Release 11, Update 1). High-quality sequences were obtained using the ‘Quality’ option in the RDP database. Search terms used were ‘cow feces’, ‘cow faeces’, ‘cattle feces’, ‘cattle faeces’, ‘fecal cow’ and ‘fecal cattle,’ and then some sequences that did not originate from cattle feces were manually removed. In addition, 16S rRNA gene sequences of bacteria isolated from cattle feces were manually obtained from American Type Culture Collection (ATCC), Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH (DSMZ), and Japan Collection of Microorganisms (JCM). The collective sequence dataset and the navigation tree were downloaded from the RDP and then imported into the ARB program [11] to construct the taxonomic tree with the Bergey’s taxonomy [23].
Diversity Estimate
The QIIME software package 1.6.0 [2] was used to cluster OTUs against a Greengenes reference set (2013-05) using the closed reference OTU picking method and calculate diversity indices [12, 24]. The maximum number of OTUs was estimated from a rarefaction curve using the nonlinear model procedure (PROC NLIN) of SAS (V9.2, SAS Institute Inc., Cary, NC) as described previously [10].
OTUs Shared Between the Rumen and the Feces
OTUs shared between the rumen and the feces were analyzed using the QIIME software package 1.6.0 [2]. For this analysis, we used the ruminal sequences that were retrieved from the RDP database previously [10].
Results and Discussion
The current study provided a global view of the fecal bacterial diversity analyzed from collective 13,663 sequences that were publicly available in the RDP database (Table 1). The 13,663 sequences were retrieved from 19 published studies and 9 unpublished studies conducted using the Sanger sequencing technology, where 603 sequences were recovered from bacterial isolates. 11,048 of the 13,663 sequences were obtained from the feces of heifers fed 61.6 % corn silage, 15.2 % alfalfa hay, 20.9 % corn, and 2.3 % liquid supplement ([4]; designated as “Dataset 1”), while 566 of the 603 isolate sequences were obtained from 8-week continuous culture enrichments of bovine fecal bacteria with cellulose or xylan/pectin ([27]; designated as “Dataset 2”).
Data Summary
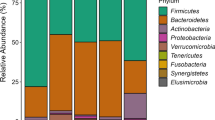
13,447 of the 13,663 sequences were assigned to 10 phyla, 17 classes, 28 orders, 59 families, and 110 genera, while the remaining 216 sequences could not be assigned to a known phylum (Table 1). Firmicutes and Bacteroidetes were the first and the second largest phyla and accounted for 49 and 42 % of the total sequences, respectively. The predominance of these two phyla in cattle feces corroborated the previous studies using the next-generation pyrosequencing technology [8, 17, 19]. Therefore, these two phyla might play more important role in the fecal microbial ecosystem than the other phyla. Proteobacteria was the third largest phylum and accounted for 6 % of the total sequences, while the remaining 7 minor phyla each accounted for <0.6 % of the total sequences.
Firmicutes
Firmicutes was represented by 6697 sequences, where 314 sequences were recovered from bacterial isolates (Table 1). 299 of the 314 isolate sequences were recovered from Dataset 2. Clostridia was the largest class and comprised 5564 of the 6697 sequences, followed by Erysipelotrichia (665 sequences), Negativicutes (219 sequences), and Bacilli (175 sequences). 49 known genera and 9 putative genera were identified within Firmicutes.
Faecalibacterium was the largest genus and comprised 810 sequences, where 809 sequences from Dataset 1 were recovered from the feces of cattle fed a diet including 21 % corn [4]. Kim et al. [8] indicated that the abundance of Faecalibacterium was high in the feces of cattle fed a corn-based diet but very low in the feces of cattle fed a mostly forage diet. Therefore, Faecalibacterium might be commonly found in the feces of cattle fed a corn-based diet but not in the feces of cattle fed a mostly forage diet. Faecalibacterium might play a role in producing butyrate, the main energy source for the gut epithelial cells [16], in cattle fed a corn-based diet. Blautia was the second largest genus and comprised 362 sequences, where 334 sequences were recovered from Dataset 1, while 27 isolate sequences were recovered from continuous culture enrichments of bovine fecal bacteria with xylan/pectin (Dataset 2). Although the abundance of Blautia was greater in the feces of cattle fed a corn-based diet than in the feces of cattle fed a mostly forage diet [8], some Blautia spp. corresponding to the 27 isolate sequences may be involved in fiber digestion. Roseburia was the third largest genus and comprised 267 sequences. Because Roseburia sequences were mostly recovered from Dataset 1 (264 sequences), it is thought to be abundant in the feces of cattle fed diets with grain. This result supported the previous study [8], where the abundance of Roseburia was higher in the feces of cattle fed a corn-based diet but very low in the feces of cattle fed a mostly forage diet. Roseburia can produce butyrate like Faecalibacterium [16].
Bacillus was represented by only 9 sequences but included 5 isolate sequences (Table 1). Bacillus pumilus, Bacillus circulans, Bacillus subtilis, Bacillus pseudomycoides, and Bacillus megaterium were each represented by one isolate sequence in an unpublished study. Kim et al. [8] also indicated that Bacillus was not a dominant genus in both corn-fed and mostly forage-fed animals. Therefore, Bacillus might be a minor genus in cattle feces irrelevant to diet but seemed to be easily enriched and isolated. Paenibacillus was represented by 3 isolate sequences, where 2 sequences were recovered from Paenibacillus favisporus that is xylanolytic [22] and the other sequence was recovered from continuous culture enrichments of bovine fecal bacteria with xylan/pectin (Dataset 2). This result indicated that Paenibacillus spp. in cattle feces might be involved in fiber digestion. Lactobacillus producing lactic acid was represented by 17 sequences including 2 isolate sequences. One isolate sequence was recovered from Lactobacillus amylovorus, while another isolate sequence was recovered from Lactobacillus johnsonii. The remaining 15 Lactobacillus sequences were recovered from Dataset 1. Streptococcus was represented by 119 sequences, where 2 sequences were recovered from Streptococcus bovis. Lachnobacterium producing lactic acid was represented by 2 isolate sequences recovered from Lachnobacterium bovis isolated from the feces of steers fed 50 % rolled barley and 50 % chopped alfalfa [25], while Weissella was represented by 2 isolate sequences recovered from Weissella spp. in an unpublished study. Because Weissella is a lactic acid bacterium associated with food [21], Weissella spp. in cattle feces also might produce lactic acid. The putative genus Clostridium XIVa comprised 88 sequences including 73 isolate sequences recovered from Dataset 2 (Table 1), indicating that Clostridium XIVa may be involved in fiber digestion. Some sequences recovered from Dataset 2 could not be assigned to a known genus, indicating that unknown genera may play a role in degrading ruminally undigested fibers in cattle feces. The current taxonomy will need to be updated to better understand the diversity of bovine fecal bacteria.
Bacteroidetes
Bacteroidetes comprised 5691 sequences, where 5368 sequences were assigned to the class Bacteroidia in which 17 known genera were identified (Table 1). The remaining 323 sequences could not be assigned to a known class.
Prevotella was the largest genus and comprised 3026 sequences (22.1 % of the total sequences) but was represented by no isolate sequences. 2995 of the 3026 sequences were recovered from Dataset 1, which indicated that Prevotella is commonly found in the feces of cattle fed a corn-based diet as described previously [8]. Bacteroides was the second largest genus and comprised 765 sequences, where 538 sequences were recovered from Dataset 1, while 121 sequences were recovered from Dataset 2. Bacteroides spp. corresponding to Dataset 1 might be commonly found in the feces of cattle fed a corn-based diet, while Bacteroides spp. corresponding to Dataset 2 may play a role in degrading ruminally undigested fibers in cattle feces. Durso et al. [5] noted that Prevotella increased in the feces of cattle fed a standard corn-based diet, while Bacteroides increased in the feces of cattle fed a diet of 40 % corn-based wet distillers’ grains with solubles. Although Prevotella and Bacteroides are commonly found in the feces of cattle fed a corn-based diet, they seem to be differently affected by dietary sources. Paraprevotella were the third largest genus and comprised 358 sequences. Paraprevotella, which produces succinate and acetate, is a novel genus placed within the family Prevotellaceae but is phylogenetically distinct (>12 % sequence dissimilarity) from Prevotella placed within the same Prevotellaceae [13]. Parabacteroides, which is a novel genus differentiated from Bacteroides [18], was the fourth largest genus and comprised 119 sequences. 15 of the 119 Parabacteroides sequences were recovered from Dataset 2, indicating that some Parabacteroides spp. may degrade ruminally undigested fibers in cattle feces. Dysgonomonas and Proteiniphilum were represented by 32 and 13 isolate sequences only recovered from Dataset 2, respectively, which indicated that these two genera may play a role in degrading ruminally undigested fibers in cattle feces. The rest of the genera placed within Bacteroidetes were represented by no isolate sequences.
Proteobacteria
Proteobacteria comprised 888 sequences including 69 isolate sequences (Table 1). Gammaproteobacteria was the largest class and comprised 717 of the 888 sequences. A total of 26 known genera were identified from the proteobacterial sequences. Succinivibrio was the largest genus and comprised 506 sequences, where 503 sequences were recovered from Dataset 1. Succinivibrio seems to be abundant in a corn-based diet as described previously [8]. Pseudomonas was the second largest genus and comprised 104 sequences. Campylobacter was represented by 9 isolate sequences, where 4 sequences were recovered from Campylobacter hyointestinalis and one sequence was recovered from Campylobacter sputorum. Arcobacter was represented by one isolate sequence recovered from Arcobacter skirrowii, while Escherichia/Shigella was represented by 20 sequences including 2 isolate sequences recovered from Escherichia coli. Proteus (31 sequences), Desulfovibrio (18 sequences), Aeromonas (7 sequences), Morganella (1 sequence), and Citrobacter (1 sequence) were represented by sequences only recovered from Dataset 2, indicating that these genera may play a role in degrading ruminally undigested fibers in cattle feces.
Minor Phyla
Actinobacteria comprised 79 sequences including 31 isolate sequences (Table 1). A total of 11 known genera were identified, where 9 genera were each represented by at least one isolate sequence. Bifidobacterium was represented by 30 sequences that were assigned to 6 OTUs. One OTU was represented by 2 sequences recovered from Bifidobacterium longum. Another one OTU was represented by sequences recovered from continuous culture enrichments of bovine fecal bacteria with xylan/pectin (Dataset 2). Species corresponding to this OTU may be related to Bf. pseudolongum that can ferment pectin [20], which may be predominant in cattle feces as described previously [6]. Further study will need to be conducted to corroborate this observation. Mycobacterium was represented by 3 isolate sequences recovered from Mycobacterium avium, while Rhodococcus was represented by 2 isolate sequences recovered from Rhodococcus coprophilus. Propionibacterium (5 sequences), Olsenella (1 sequence), and Paraeggerthella (1 sequence) were recovered from Dataset 2, indicating that these genera may be involved in fiber digestion. Amycolatopsis (1 sequence), Micromonospora (2 sequences), and Streptomyces (2 sequences) were represented by isolate sequences recovered from another unpublished study.
Spirochaetes comprised 47 sequences including no isolate sequences, and all 47 sequences were assigned to the genus Treponema (Table 1). The 47 sequences were recovered from either dairy cattle in Korea [7] or Holstein cattle in the USA [15], but neither study provided diet information. Tenericutes comprised 27 sequences, where 24 sequences recovered from Dataset 1 were assigned to the genus Anaeroplasma. Synergistetes comprised 8 sequences that were assigned to the genus Cloacibacillus, while Fusobacteria comprised 7 sequences that were assigned to the genus Fusobacterium. Both Cloacibacillus and Fusobacterium were represented by isolate sequences recovered from continuous culture enrichments of bovine fecal bacteria with xylan/pectin (Dataset 2), which indicated that they may degrade ruminally undigested fibers in cattle feces. Verrucomicrobia was represented by only 2 sequences, while Elusimicrobia was represented by only one sequence. Fecal bacteria in these two phyla might not be residents in cattle feces.
Bacterial Diversity
A total of 1252 OTUs at 97 % sequence similarity were identified from the total sequences using the closed reference OTU picking method (Table 1). The number of OTUs identified using the open reference OTU picking method was 1109 that were excluded from OTU calculation because of some non-overlapping sequences. The 1109 OTUs indicate novel bacterial species. The OTUs represented by only one sequence accounted for 52 % of all 1252 OTUs, indicating that fecal bacterial communities of cattle are greatly diverse. Although 80 % of the total sequences were recovered from Dataset 1, the number of OTUs shared between Dataset 1 and the other datasets was only 2 of all 1252 OTUs. This result supports the finding that the composition of bovine fecal bacteria is affected by various factors, particularly diet, as described previously [8]. Therefore, small-scale studies that were conducted under different experimental conditions still help find novel OTUs [9].
The maximum OTU richness predicted by the rarefaction curve was 1494, indicating that more than 80 % of fecal bacterial diversity has been sampled. However, actual % coverage of fecal bacterial diversity will be much lower than 80 % because this analysis did not include novel OTUs identified using the open reference OTU picking method.
Only 82 of the 1252 OTUs were shared between the rumen and the feces (Fig. 1). This result indicated that the community structure of fecal bacteria is distinct from that of the rumen bacteria. The 82 shared OTUs were assigned to Anaerorhabdus (1 OTU), Anaerovorax (1 OTU), Bacteroides (1 OTU), Bifidobacterium (3 OTUs), Blautia (1 OTU), Campylobacter (1 OTU), Clostridium sensus stricto (2 OTUs), Clostridium XIVa (2 OTUs), Desulfovibrio (1 OTU), Enterococcus (2 OTUs), Escherichia/Shigella (2 OTUs), Fusobacterium (2 OTUs), Lachnobacterium (1 OTU), Lachnospiraceae incertae sedis (1 OTU), Lactobacillus (3 OTUs), Lactococcus (1 OTU), Mogibacterium (3 OTUs), Olsenella (2 OTUs), Prevotella (6 OTUs), Proteiniphilum (1 OTU), Proteus (1 OTU), Ruminococcus (4 OTUs), Selenomonas (1 OTU), Sharpea (1 OTU), Sphingomonas (1 OTU) , Sporanaerobacter (1 OTU), Streptococcus (1 OTU), Subdivision 5 genera incertae sedis (1 OTU), Syntrophococcus (2 OTUs), Tissierella (1 OTU), Treponema (1 OTU), and taxa that could not be assigned to a known genus (30 OTUs).

A venn diagram showing OTUs shared between the rumen and the feces. The number of the shared OTUs was 82 OTUs, accounting for only 6.5 % of the 1252 fecal OTUs
In conclusion, the current study provided a collective view of fecal bacterial communities using a meta-analysis of studies based on the Sanger sequencing technology and indicated that individual small-scale studies can help better understand fecal bacterial communities of cattle. Sequences recovered from cultured bacteria accounted for only 4 % of the total sequences. Therefore, more efforts would need to be made to isolate and characterize fecal bacteria.
References
Callaway TR, Dowd SE, Edrington TS, Anderson RC, Krueger N, Bauer N, Kononoff PJ, Nisbet DJ (2010) Evaluation of bacterial diversity in the rumen and feces of cattle fed different levels of dried distillers grains plus solubles using bacterial tag-encoded FLX amplicon pyrosequencing. J Anim Sci 88:3977–3983
Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R (2010) QIIME allows analysis of high-throughput community sequencing data. Nat Methods 7:335–336
Dowd SE, Callaway TR, Wolcott RD, Sun Y, McKeehan T, Hagevoort RG, Edrington TS (2008) Evaluation of the bacterial diversity in the feces of cattle using 16S rDNA bacterial tag-encoded FLX amplicon pyrosequencing (bTEFAP). BMC Microbiol 8:125
Durso LM, Harhay GP, Smith TP, Bono JL, Desantis TZ, Harhay DM, Andersen GL, Keen JE, Laegreid WW, Clawson ML (2010) Animal-to-animal variation in fecal microbial diversity among beef cattle. Appl Environ Microbiol 76:4858–4862
Durso LM, Wells JE, Harhay GP, Rice WC, Kuehn L, Bono JL, Shackelford S, Wheeler T, Smith TP (2012) Comparison of bacterial communities in faeces of beef cattle fed diets containing corn and wet distillers’ grain with solubles. Lett Appl Microbiol 55:109–114
Gavini F, Delcenserie V, Kopeinig K, Pollinger S, Beerens H, Bonaparte C, Upmann M (2006) Bifidobacterium species isolated from animal feces and from beef and pork meat. J Food Prot 69:871–877
Jeong JY, Park HD, Lee KH, Weon HY, Ka JO (2011) Microbial community analysis and identification of alternative host-specific fecal indicators in fecal and river water samples using pyrosequencing. J Microbiol 49:585–594
Kim M, Kim J, Kuehn L, Bono JL, Berry ED, Kalchayanand N, Freetly HC, Benson AK, Wells JE (2014) Investigation of bacterial diversity in the feces of cattle fed different diets. J Anim Sci 92:683–694
Kim M, Morrison M, Yu Z (2011) Phylogenetic diversity of bacterial communities in bovine rumen as affected by diets and microenvironments. Folia Microbiol (Praha) 56:453–458
Kim M, Morrison M, Yu Z (2011) Status of the phylogenetic diversity census of ruminal microbiomes. FEMS Microbiol Ecol 76:49–63
Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar Buchner A, Lai T, Steppi S, Jobb G, Förster W, Brettske I, Gerber S, Ginhart AW, Gross O, Grumann S, Hermann S, Jost R, König A, Liss T, Lüssmann R, May M, Nonhoff B, Reichel B, Strehlow R, Stamatakis A, Stuckmann N, Vilbig A, Lenke M, Ludwig T, Bode A, Schleifer KH (2004) ARB: a software environment for sequence data. Nucleic Acids Res 32:1363–1371
McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P (2012) An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 6:610–618
Morotomi M, Nagai F, Sakon H, Tanaka R (2009) Paraprevotella clara gen. nov., sp. nov. and Paraprevotella xylaniphila sp. nov., members of the family ‘Prevotellaceae’ isolated from human faeces. Int J Syst Evol Microbiol 59:1895–1900
Ozutsumi Y, Hayashi H, Sakamoto M, Itabashi H, Benno Y (2005) Culture-independent analysis of fecal microbiota in cattle. Biosci Biotechnol Biochem 69:1793–1797
Patton TG, Scupham AJ, Bearson SM, Carlson SA (2009) Characterization of fecal microbiota from a Salmonella endemic cattle herd as determined by oligonucleotide fingerprinting of rDNA genes. Vet Microbiol 136:285–292
Pryde SE, Duncan SH, Hold GL, Stewart CS, Flint HJ (2002) The microbiology of butyrate formation in the human colon. FEMS Microbiol Lett 217:133–139
Rice WC, Galyean ML, Cox SB, Dowd SE, Cole NA (2012) Influence of wet distillers grains diets on beef cattle fecal bacterial community structure. BMC Microbiol 12:25
Sakamoto M, Benno Y (2006) Reclassification of Bacteroides distasonis, Bacteroides goldsteinii and Bacteroides merdae as Parabacteroides distasonis gen. nov., comb. nov., Parabacteroides goldsteinii comb. nov. and Parabacteroides merdae comb. nov. Int J Syst Evol Microbiol 56:1599–1605
Shanks OC, Kelty CA, Archibeque S, Jenkins M, Newton RJ, McLellan SL, Huse SM, Sogin ML (2011) Community structures of fecal bacteria in cattle from different animal feeding operations. Appl Environ Microbiol 77:2992–3001
Slovakova L, Duskova D, Marounek M (2002) Fermentation of pectin and glucose, and activity of pectin-degrading enzymes in the rabbit caecal bacterium Bifidobacterium pseudolongum. Lett Appl Microbiol 35:126–130
Stiles ME, Holzapfel WH (1997) Lactic acid bacteria of foods and their current taxonomy. Int J Food Microbiol 36:1–29
Velazquez E, de Miguel T, Poza M, Rivas R, Rossello-Mora R, Villa TG (2004) Paenibacillus favisporus sp. nov., a xylanolytic bacterium isolated from cow faeces. Int J Syst Evol Microbiol 54:59–64
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol 73:5261–5267
Werner JJ, Koren O, Hugenholtz P, DeSantis TZ, Walters WA, Caporaso JG, Angenent LT, Knight R, Ley RE (2012) Impact of training sets on classification of high-throughput bacterial 16 s rRNA gene surveys. ISME J 6:94–103
Whitford MF, Yanke LJ, Forster RJ, Teather RM (2001) Lachnobacterium bovis gen. nov., sp. nov., a novel bacterium isolated from the rumen and faeces of cattle. Int J Syst Evol Microbiol 51:1977–1981
Woese CR, Gutell R, Gupta R, Noller HF (1983) Detailed analysis of the higher-order structure of 16S-like ribosomal ribonucleic acids. Microbiol Rev 47:621–669
Ziemer CJ (2014) Newly cultured bacteria with broad diversity isolated from 8 week continuous culture enrichments of cow feces on complex polysaccharides. Appl Environ Microbiol 80:574–585
Acknowledgment
No funding sources declared.
Author information
Authors and Affiliations
Corresponding author
Additional information
Mention of a trade name, proprietary product, or specific equipment does not constitute a guarantee or warranty by the USDA and does not imply approval to the exclusion of other products that may be suitable.
USDA is an equal opportunity provider and employer.
Rights and permissions
About this article

Cite this article
Kim, M., Wells, J.E. A Meta-analysis of Bacterial Diversity in the Feces of Cattle. Curr Microbiol 72, 145–151 (2016). https://doi.org/10.1007/s00284-015-0931-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-015-0931-6