
Abstract
A 4.20-kb SspI fragment from Bacillus thuringiensis G03 was cloned and sequenced. Sequencing analysis revealed two complete open reading frames (ORF; tzw1 and tzw2), and one incomplete ORF (tzw3) (GenBank accession no. EU293887). Tzw1 encodes a putative nonribosomal peptide synthetase with thiolation and condensation domains localized at the C-termini, whereas tzw2 and tzw3 encode acyl carrier protein and Acyl-CoA dehydrogenase, respectively. To investigate the function of tzw1 in zwittermicin A (ZA) biosynthesis, an in-frame deletion of 1,461 bp within tzw1 was constructed. The mutant abolished ZA production. Complementation of the mutant with cloned tzw1 restored ZA productivity. These results revealed that tzw1 is required for ZA biosynthesis in B. thuringiensis G03.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
B. thuringiensis, the well-known and widely used insecticidal agent, is a ubiquitous Gram-positive, rod-shaped, spore-forming bacterium. The significant insecticidal activity mostly owes to the parasporal crystal proteins formed during the stationary phase of its growth [8]. Zwittermycin (ZA) (Fig. 1A), a novel aminopolyol compound recently isolated from the fermentation broths of B. thuringiensis and B. cereus, acts synergistically with the crystal proteins to enhance the insecticidal activity of B. thuringiensis [1]. It also has broad-spectrum antibiotic activity against oomycetes, algae protists, many plant pathogenic fungi, and certain bacteria [10].

Zwittermicin A and gene organization of the 4.24-kb sequenced region. (A) Structure of zwittermicin A. (B) Gene organization of the 4.24-kb sequenced region. T, thiolation domain; C, condensation domain; dotted arrow, incomplete ORF; ATGA, overlapped stop codon of tzw2 and start codon of tzw3
Handelsman et al. previously identified the ZA self-resistance gene (zmaR) and other three genes involved in biosynthesis that are adjacent to zmaR through whole-genome random cloning and transposon mutagenesis [6, 12]. Extensive biochemical assays established that ZmaR shows acetyltransferase activity to inactivate ZA by acetylation modification [13]. Subsequently, a 16-kb DNA fragment flanking the reported four genes was cloned and sequenced. Another five genes and one partial gene were identified to be required for ZA biosynthesis. Most noteworthy is the presence of nonribosomal peptide synthetase (NRPS) and polyketide synthase (PKS) genes, suggesting that ZA is synthesized by NRPS–PKS machinery, which is in strong agreement with its chemical features [4]. Recently, genes responsible for the biosynthesis of hydroxymalonyl-ACP and aminomalonyl-ACP extender units were biochemically characterized [2]. Sun et al also localized three genes possibly responsible for the carbamoylation or biosynthesis of the putative 2, 3-diaminopropionate extender unit [15].
So far, some other previously identified essential genes, including NRPSs, have not been localized, and other essential genes, such as aminotransferase gene, are still missing. Therefore, it is necessary to clone the entire ZA biosynthetic gene cluster and identify more required genes. Herein we report the cloning of a region related to ZA biosynthesis and characterization of the NRPS gene tzw1.
Materials and Methods
Bacterial Strains, Plasmids, and Culture Conditions
B. thuringensis G03 is the wild-type ZA producer [9]. Erwinia herbicola OS, the indicator for ZA bioassay, was kindly provided by Kernel Bio-Pesticide Company (Wuhan, PRC). Escherichia coli JM110 (Merck) is used as gene cloning host, whereas E. coli SCS110 (Promega) is used to obtain nonmethylated plasmid DNA. B. thuringensis–E. coli shuttle thermosensitive vector pRN5101 was kindly provided by D. Lereclus and used for gene inactivation. B. thuringensis–E. coli shuttle plasmid pSTK was used for gene complementation [14]. Plasmid pBAC-TZW01 from the bacterial artificial chromosome (BAC) genomic library of B. thuringensis G03 has a 90-kb insert containing the ZA biosynthetic gene cluster (Shao et al., unpublished).
Brain–heart medium (BD 237500) was used for preparation of B. thuringiensis competent cells. The fermentation medium for ZA production contained 3 g−1 beef extract and 5 g l−1 peptone (pH 7.2). All B. thuringiensis strains were grown in Lurai-Bertani (LB) broth at 28°C for 12 hours as seed cultures, which were then diluted 1/100 in 20 mL fermentation medium incubated at 28°C and 220 rpm for 3 days. E. coli and B. thuringiensis were cultivated at 37°C and 28°C, respectively, in LB broth or on LB agar plates [7]. For screening of the mutants with double cross-over recombination, 39°C was used for the first round of growth, and 42°C was used for the second round of growth. All of the antibiotics were purchased from Sigma, and the final concentrations were 50 μg mL−1 kanamycin, 100 μg mL−1 ampicillin, and 10 μg mL−1 erythromycin.
DNA Manipulations
Extraction of chromosomal DNA of B. thuringiensis was performed according to established techniques [3]. B. thuringiensis competent cells were prepared, and electroporation was carried out according to Lereclus [5]. Plasmid isolation from E. coli, transformation of E. coli, and Southern blotting were manipulated as previously described [7]. Digoxin labeling and detection kits (Boehringer Mannheim) were used according to protocols of the manufacturer. Primer synthesis and DNA sequencing were performed by Shanghai Sangon. Restriction enzymes and other modifying enzymes were obtained from TaKaRa.
Construction of the tzw1 Mutant
Overlap extension polymerase chain reaction (PCR) with KOD-Plus (Toyobo) was carried out for construction of the tzw1 mutant. Total DNA of B. thuringiensis G03 was used as the template for the amplification. Primers of PMC1 (5′-CGC-GGATCCGCTTTCCCTCTTAGCTTTCTG-3′ [with BamHI site underlined]) and PMC2 (5′-CTTTACAG-GTGGCC TGAAACAGTCTTCTTC-3′ [with nucleotides from the upstream underlined and nucleotides from the downstream in bold]) were used for amplification of the 671-bp left-flanking region. Primers of PMC3 (5′-GAAGAAGACTGTTTCAGGCCACCTGTAAAG-3′ [exactly complementary with PMC2]) and PMC4 (5′-CGCCAGCTGGCAACAATAGTTTCCTCTGC-3′ [with PvuII site underlined]) were used for amplification of the 871-bp right-flanking region. After mixing the 671- and 871-bp amplified products as templates, overlap extension PCR was performed. The resulting 1.50-kb product was inserted into pRN5101 to generate pRN5101-tzw1. The insert was confirmed by sequencing.
Nonmethylated pRN5101-tzw1 was obtained from E. coli SCS110 and then introduced into B. thuringiensis G03 by way of electroporation. The transformants were selected on LB agar plates containing 10 μg mL−1 erythromycin. One transformant was cultivated for 10 hours at a nonpermissive temperature of 39°C without antibiotic selection. After plating and growing on an erythromycin-containing plate, one erythromycin-resistant colony was cultivated for another 10 hours at a nonpermissive temperature of 42°C without antibiotic selection. Collected cells were plated on antibiotic-free plates and screened for erythromycin-sensitive derivatives. Erythromycin-sensitive colonies were characterized initially through PCR amplification with primers of PMC1 and PMC4 and subsequently by Southern hybridization against EcoRI-digested total DNA using the 470-bp probe amplified with primers SBCF (5′-GAGTTTTGAGGGTGAAC-3′) and SBCR (5′-CTGTGTTTGTTTCCGTGC-3′).
Complementation of the tzw1 Mutant
Intact tzw1 was amplified with the oligonucleotide primers of PCCF (5′-CGCGGATCCGATGGGAAATTATACTTCTTCAATC-3′ [with BamHI site underlined]) and pCCR (5′-ACGCGTCGACTTAACCTATATCCCAATTAAAATC-3′ [with SalI site underlined]). The 2.65-kb amplified product was digested with BamHI and SalI and cloned into pSTK to generate pSTK-tzw1. After sequence confirmation of the insert, pSTK-tzw1 was then introduced into the Δtzw1 mutant by way of electroporation.
Bioassay of ZA
Bioassay was performed as described [11] with minor modification using 0.5 × LB as indicating medium.
Liquid Chromatography–Mass Spectrometry Analysis
Liquid chromatography–mass spectrometry (LC-MS) was carried out using a Waters series high-pressure liquid chromatographer 2695 (Waters) with a Thermo Finnigan LCQ Advantage (Thermo Finnigan) mass detector (ion trap). The instrument conditions were optimized as follows: spray voltage 5.2 kV, sheath gas flow rate 1.50 Mpa, capillary voltage 47.00 V, capillary temperature 250°C, and normalized collision energy 29% to approximately 35%. The samples were separated by an Xterra PR-18 5-μm pore size 3.9 × 150-mm column (Waters) with isocratic elution (40% methanol and 0.1% [v/v] formic acid) at a flow rate of 100 μL min−1. ZA standard was kindly provided by Kernel Bio-Pesticide Company (Wuhan, PRC).
Results
Sequencing of a 4,243-bp Fragment in ZA Biosynthetic Gene Cluster
A 4,243-bp SspI DNA fragment was subcloned from pBAC-TZW01, a positive BAC plasmid containing ZA biosynthetic cluster (Shao et al., unpublished) and sequenced. Two complete open-reading frames (ORFs; tzw1 and tzw2) and one incomplete ORF (tzw3) were revealed in this fragment. The distance between tzw1 and tzw2 was 33 bp, whereas tzw2 and tzw3 overlapped for 4 bp, suggesting that all three genes transcribed as one operon (Fig. 1B). Tzw1, in close proximity to the 5′-terminal of the previously cloned 16-kb ZA biosynthesis cluster from B. cereus [4], encodes a putative protein with 883 amino acids showing high identity to FenD of the Fengicin biosynthetic pathway in B. amyloliquefaciens FZB42 (identities 42% and similarity 64%). The C-terminal portion of Tzw1 (346 to 859 amino acids) contains a thiolation (T) domain and a condensation (C) domain, the typical feature of NRPS enzymes.
The putative protein of tzw2 is comprised of 87 amino acids and shows 98% identity with ORF3 (ZmaD), whereas the putative 312-amio acid product of tzw3 shows 99% identity with ORF1 (ZmaE) of the ZA biosynthesis cluster from B. cereus [4], both of which were proposed to be involved in the biosynthesis of the hydroxy-malonyl-ACP extender unit [2].
Gene Inactivation of tzw1
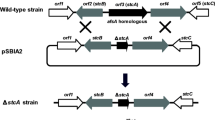
An in-frame deletion was performed to test if tzw1 is involved in ZA biosynthesis. The sequence of the pRN5101-tzw1 revealed that the 1,461-bp fragment (428 to 1,888 bp) of tzw1 was deleted (Fig. 2A). Nonmethylated pRN5101-tzw1 was transformed into B. thuringiensis G03, and disrupted mutants were constructed by way of double cross-over of homologous recombination. Mutants were firstly proved through PCR using PMC1 and PMC4 (data not shown). Using the 470-bp PCR-amplified fragment (from 1,902 to 2,371 bp) of tzw1 as the probe, the results of Southern hybridization showed the expected 2.60 and 1.20 kb in the wild-type and the tzw1 mutant (Δtzw1), respectively (Fig. 2B). In the bioassay against the indicator Erwinia herbicola OS with fermentation broths, Δtzw1 lost inhibitory activity, whereas the wild-type strain yielded a clear inhibition zone, unambiguously proving the involvement of tzw1 in the biosynthesis of ZA (Fig. 2C). Further support came from LC-MS analysis of the fermentation broths along with the purified ZA standard, in which ZA, with m/z 397 ([M + H]+) and 419 ([M + Na]+), was absent in Δtzw1 (Fig. 2D). The peaks with m/z 397 ([M + H]+) in the standard and the wild-type strains were further fragmented into the typical 251 ([M - C4H9N4O2]+) and 379 ([M + H – H2O]+) fragments (data not shown), which most probably resulted from breakage of the amide bond and from dehydration, respectively.

Gene inactivation of tzw1. (A) Schematic representation of the 1,461-bp in-frame deletion of tzw1. The total DNA was digested with EcoRI for Southern hybridization and the expected signals for wild-type and tzw1 mutant were 2618 and 1157 bp, respectively. ermE, erythromycin resistant gene; ori (ts), temperature-sensitive origin of replication; solid bar, 470-bp probe from 1,902 to 2,371 bp of tzw1. (B) Southern hybridization analysis of the wild-type and the tzw1 mutant. M, molecular-weight standard. (C) Bioassay of wild-type, mutant, and complemented strains. a, wild-type; b, tzw1 mutant (Δtzw1); c, complemented Δtzw1 with cloned tzw1. Er. herbicola OS was used as indicator strain. (D) LC-MS comparison of standard, wild-type, mutant, and complemented strains. a, standard; b, wild-type; c, tzw1 mutant (Δtzw1); d, complemented Δtzw1 with cloned tzw1. The m/z of ZA was 397.3 ([M + H]+)
Complementation of Δtzw1
To verify that the loss of ZA productivity in Δtzw1 resulted only from tzw1 inactivation, the 2.56-kb fragment containing whole-length tzw1 was amplified by PCR, then it was cloned into plasmid pSTK and introduced into the tzw1 mutant by way of electroporation. A derivative of Δtzw1 with cloned tzw1 regenerated the productivity of ZA as shown in bioassay with inhibitory activity against Er. herbicola OS (Fig. 2C) and LC-MS analysis with the presence of peak corresponding to m/z 397.3 ([M + H]+) (Fig. 2D).
Discussion
Previous work from several groups localized a 16-kb region plus three other genes involved in ZA biosynthesis [4, 15]. However, other genes, possibly required for biosyntheses of NRPS extension, PKS elongation, and transamination, are still missing. Moreover, random transposon mutagenesis generated other ZA-null mutants and sequencing of the flanking sequences of each transposon identified at least four NRPS and two PKS loci not located in the sequenced 16-kb region. The newly found NRPS gene tzw1 is most probably transcribed with tzw2 and tzw3, both of which showed high homology with orf3 and orf1 [at the left-most of the 16-kb region] from B. cereus, respectively. Although conserved T and C domains of NRPS were identified in Tzw1, no adenylation domain was eligible, and the 356-amino acid N-terminal region does not show homology with any protein. Orf8 in the ZA biosynthetic gene cluster from B. cereus is a NRPS–PKS hybrid, and domain analysis strongly suggested its involvement in the initiation with serine and with the first elongation with malonyl-CoA [4]. Subsequent elongation steps with hydroxymalonyl-ACP and aminomalonyl-ACP are supposed to be performed by PKS [2]. Therefore, Tzw1 is probably involved in the activation and incorporation of the 2,3-diaminopropionate extender unit to form the complete ZA backbone.
Our results here improve our understanding of the biosynthesis of ZA, pave the way for a large-scale sequencing of the BAC clone containing ZA biosynthetic genes, and set the stage for rational structure modification through combinatory biosynthesis.
References
Broderick NA, Goodman RM, Raffa KF, Handelsman J (2000) Synergy between zwittermicin A and Bacillus thuringiensis subsp. kurstaki against gypsy moth (Lepidoptera: Lymantriidae). Environ Entomol 29:101–107
Chan YA, Boyne 2nd MT, Podevels AM, Klimowicz AK, Handelsman J, Kelleher NL et al (2006) Hydroxymalonyl-acyl carrier protein (ACP) and aminomalonyl-ACP are two additional type I polyketide synthase extender units. Proc Natl Acad Sci U S A 103:14349–14354
Delecluse A, Charles JF, Klier A, Rapoport G (1991) Deletion by in vivo recombination shows that the 28-kilodalton cytolytic polypeptide from Bacillus thuringiensis subsp. israelensis is not essential for mosquitocidal activity. J Bacteriol 173:3374–3381
Emmert EA, Klimowicz AK, Thomas MG, Handelsman J (2004) Genetics of zwittermicin a production by Bacillus cereus. Appl Environ Microbiol 70:104–113
Lereclus D, Arantes O, Chaufaux J, Lecadet M (1989) Transformation and expression of a cloned delta-endotoxin gene in Bacillus thuringiensis. FEMS Microbiol Lett 51:211–217
Milner JL, Stohl EA, Handelsman J (1996) Zwittermicin A resistance gene from Bacillus cereus. J Bacteriol 178:4266–4272
Sambrook J, Russell DW (2001) Molecular cloning — A laboratory manual, 3rd ed. Cold Spring Harbor Laboratory Press, New York, NY
Schnepf E, Crickmore N, Van Rie J et al (1998) Bacillus thuringiensis and its pesticidal crystal proteins. Microbiol Mol Biol Rev 62:775–806
Shao TM, Song FP, Li ZF, Zhang J, Jiang SH, Liu DQ et al (2005) Screening of zwittermicin a-producing strains of Bacillus thuringiensis and cloning and expression of zmaR gene. Agric Sci China 38:1811–1816
Silo-Suh LA, Stabb EV, Raffel SJ, Handelsman J (1998) Target range of zwittermicin A, an aminopolyol antibiotic from Bacillus cereus. Curr Microbiol 37:6–11
Silo-Suh LA, Lethbridge BJ, Raffel SJ, He H, Clardy J, Handelsman J (1994) Biological activities of two fungistatic antibiotics produced by Bacillus cereus UW85. Appl Environ Microbiol 60:2023–2030
Stohl EA, Milner JL, Handelsman J (1999) Zwittermicin A biosynthetic cluster. Gene 237:403–411
Stohl EA, Brady SF, Clardy J, Handelsman J (1999) ZmaR, a novel and widespread antibiotic resistance determinant that acetylates zwittermicin A. J Bacteriol 181:5455–5460
Wang G, Zhang J, Song F, Wu J, Feng S, Huang D (2006) Engineered Bacillus thuringiensis GO33A with broad insecticidal activity against lepidopteran and coleopteran pests. Appl Microbiol Biotechnol 72:924–930
Zhao C, Luo Y, Song C, Liu Z, Chen S, Yu Z et al (2007) Identification of three Zwittermicin A biosynthesis-related genes from Bacillus thuringiensis subsp. kurstaki strain YBT-1520. Arch Microbiol 187:313–319
Acknowledgments
This work was supported by grants from National 973 Plan Program of China (Grant No. 2003CB114201) and grants from National 863 Plan Program of China (Grant Nos. 2006AA02Z189 and 2006AA10A212).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shao, T., Bai, L., Zhang, J. et al. A Nonribosomal Peptide Synthetase Gene tzw1 Is Involved in Zwittermicin A Biosynthesis in Bacillus thuringiensis G03. Curr Microbiol 57, 61–65 (2008). https://doi.org/10.1007/s00284-008-9153-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-008-9153-5