
Abstract
Polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) analysis of amplified DNA fragment of the 16S and 23S rRNA genes was performed on 35 Helicobacter, 24 Campylobacter, and 15 Arcobacter strains. PCR amplification generated a 1004-bp fragment of 16S rDNA and a 2.6-Kbp fragment of 23S rDNA from each strain. The amplicons were digested with DdeI and HpaII, respectively. For both assays, distinctive profiles were obtained for each genus. 23S rDNA PCR-RFLP analysis with HpaII enzyme identified Campylobacter and Helicobacter strains at the species level. Analysis of 16S rRNA gene with DdeI enzyme was not useful for the specific identification of Campylobacter and Arcobacter, although it discriminated among Helicobacter species. The PCR-RFLP technique allowed for the discrimination among these three related genus with only one restriction enzyme; therefore it can be a simple, rapid, and useful method for routine identification.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The genera Helicobacter, Campylobacter, and Arcobacter form a phylogenetically distinct group included in the epsilon-subdivision of proteobacteria [36]. These spiral-shaped, Gram-negative microorganisms are microaerophilic and fastidious in culture media [38].
Identification of these bacteria has become increasingly important because many of them are now recognized as human and/or animal pathogens [27]. Thermophilic campylobacters, particularly C. jejuni and C. coli, are the most isolated bacteria causing diarrhoeal diseases in humans [12]. Foods of animal origin and drinking water are widely regarded as the main source of food-borne infection because of the presence of those organisms as part of the intestinal flora of many animals [30]. Currently, three Arcobacter species—including A. butzleri, A. cryaerophilus, and A. skirrowii—are considered to be pathogenic for domestic animals and humans [21]. The route of transmission from animals to humans is not clear, but the bacterium has been isolated from meat products and water, suggesting that Arcobacter is a possible food-borne pathogen [23].
Helicobacter genus includes a group of organisms that colonize the mucus layer covering the epithelial surface of the gastrointestinal tract of humans and a variety of animals. An important number of Helicobacter species have been related to human and animal diseases, with H. pylori being the most notable. It is the main causative agent of chronic superficial gastritis and peptic ulcer disease; it is also a risk factor for gastric cancer [5] and has been designated as a class I carcinogen by the World Health Organization [18]. H. pylori infection occurs worldwide at a high prevalence rate, and it can be eradicated in up to 90% of patients using current triple therapies, of which clarithromycin and metronidazole antibiotics are key components [6].
A growing number of enterohepatic Helicobacter species are being reported to be associated with diseases in humans [10, 29, 33]. H. pullorum has been shown to occur in poultry as well as in humans in association with gastroenteritis. This organism is especially difficult to differentiate from Campylobacter species [25]. H. fennelliae is closely related to H. pullorum (95.9% sequence similarity of 16S rRNA gene; Shen et al. [32]) and has been isolated from rectal swabs and blood from homosexual men [14]. H. fennelliae may cause gastroenteritis, cellulitis, septic arthritis, and bacteremia, most commonly in patients infected with human immunodeficiency virus (HIV) [37].
The differentiation of Helicobacter species from each other and from Campylobacter and Arcobacter species by phenotypic analysis is difficult because of the lack of standardized procedures, the well-known biochemical inertness of these organisms, the great number of cross-species phenotypic similarities, and the prevalence of atypical strains. These difficulties increase the interest in molecular approaches to identification [28].
During past decades, several molecular techniques have been applied to identify these bacteria [28]. Polymerase chain reaction–restriction fragment length polymorphism (PCR-RLFP) analysis of amplified rDNA fragments is one of the most frequent methods used to differentiate among Helicobacter, Campylobacter, and Arcobacter species [3, 20, 25] because it is more discriminatory, faster, and more cost-effective than phenotypic tests [26]. rDNA molecules present conserved regions coexisting with variable sequences that make them suitable targets for molecular identification methods. However, most of the PCR-RFLP identification schemes developed in previous works are too complex for routine use because they are unable to distinguish C. jejuni from C. coli in one step [24] or because they need more than one restriction enzyme to discriminate among Helicobacter, Campylobacter, and Arcobacter species [15]. Moreover, most of them have not been simultaneously applied to the identification of the three genera [8, 16, 19, 34]. Therefore, the objective of this study was the development of a rapid and easy-to-perform identification system based on PCR-RFLP analysis with the ability to differentiate among Helicobacter, Campylobacter, and Arcobacter species.
Material and Methods
Bacterial strains and culture conditions
A total of 74 strains were used in this study, including 31 Helicobacter strains isolated from gastric biopsies, 20 Campylobacter strains, and 12 Arcobacter strains isolated from poultry and water samples; 11 reference strains were provided by the National Collection of Type Cultures (NCTC; London, UK): H. pylori NCTC 11637, H. pylori NCTC 11638, H. pullorum NCTC 13153, H. fennelliae NCTC 11612, C. jejuni NCTC 11828, C. jejuni NCTC 12521, C. upsaliensis NCTC 11540, C. coli NCTC 11366, A. butzleri NCTC 12481, A. cryaerophilus NCTC 11885, and A. skirrowii NCTC 12713.
Helicobacter strains were grown on Columbia agar base supplemented with 10% (vol/vol) defibrinated horse blood (Oxoid; Basingstoke, UK); Arcobacter and Campylobacter were cultured on blood agar base no. 2 containing 5% (vol/vol) defibrinated sheep blood (Oxoid). Agar plates were incubated at 37°C under microaerophilic conditions (10% carbon dioxide, 5% oxygen, and 85% nitrogen) for 48 to 72 hours, except for Arcobacter, which was incubated in an aerobic atmosphere for 48 hours at 30°C. Cultures were preserved at −80°C in nutrient broth (Merck; Darmstadt, Germany) containing 10% (vol/vol) glycerol.
Biochemical characterization
Helicobacter strains isolated from biopsy specimens were characterized by colony morphology, Gram staining reaction, urease (Microkit S.L.; Madrid, Spain), catalase, and oxidase (Bactident oxidase Kit; Merck) activities. Campylobacter and Arcobacter were characterized by morphology, Gram stain, and API-Campy (Biomérieux, France) identification profiles. Arcobacter were differentiated from campylobacters by their ability to grow under aerobic conditions.
DNA isolation and PCR identification
Bacterial strains biochemically characterized as Helicobacter, Campylobacter, and Arcobacter were identified by PCR. Chromosomal DNA was extracted by cetyltrimethyl-ammonium bromide [39].
Primers C97 (5′-GCTATGACGGGTATCC-3′) and C05 (5′-ACTTCACCCCAGTCGCTG-3′) were used to amplify a 1200-bp PCR fragment of 16S rDNA from all Helicobacter species [11]. PCR was performed in 50 μl of the mixtures containing 5 μl template DNA, 1 × PCR buffer, 1.5 mM MgCl2, 200 μM each dNTP, and 0.5 μM each primer along with 2.5 U of Taq polymerase. The temperature profile for PCR was as follows: an initial step of 5 minutes at 95°C, followed by denaturation for 1 minute at 94°C, annealing for 2 minutes at 60°C, and primer extension for 2 minutes at 72°C. After the 33rd cycle, the extension step was prolonged for 5 minutes to complete synthesis of all strands, and then the samples were kept at 4°C until analysis.
Primers HP1 (5′-CCTAACCAATTGAGCCAAGAAG-3′) and HP2 (5′-CTTTCTAACACTAACGCGCTCA-3′), which amplified a 398-bp PCR fragment of 16S rDNA specific to H. pylori [4], were also used to identify the Helicobacter strains isolated from biopsy specimens. The amplification were carried out in a volume of 50 μl with 5 μl template DNA, 1 × PCR buffer, 1.5 mM MgCl2, and 200 μM each dNTP, 0.4 μM each primer, 2 U of Taq polymerase; the mixture was then submitted to an initial denaturation at 95°C for 5 minutes, followed by 33 cycles of denaturation at 94°C for 1 minute, annealing at 58°C for 1 minute, extension at 72°C for 1 minute, and a final extension step at 72°C for 5 minutes.
PCR identification of Campylobacter species was carried out according to Fermér and Engvall [9] with THERM1 (5′-ATTCCAATACCAACATTAGT-3′) and THERM4 (5′-CTTCGCTAATG CTAACCC-3′) primers, which amplified a 491-bp fragment of 23S rDNA for Campylobacter-termotolerant species.
Simultaneous identification of C. jejuni and C. coli was performed by multiplex PCR (mPCR) with three different pairs of primers. The primers used amplified a 857-bp fragment of 16S rRNA gene for Campylobacter genus [22], a 589-bp fragment of mapA gene for C. jejuni species [35], and a 462-bp fragment of ceuE gene for C. coli species [13]. mPCR was based on the assay developed by Denis et al. [7] A final reaction volume of 30 μl was made by addition of 5 μl each sample. The PCR reaction mixture contained 0.6 U Taq polymerase, 1 × PCR buffer, 200 μM each dNTP, 1.5 mM MgCl2, 0.5 μM MD16S1 and MD16S2 primers, and 0.42 μM MDmapA1, MDmapA2, COL3, and MDCOL2. The amplification consisted of an initial denaturation step at 95°C for 10 minutes followed by 35 cycles (denaturation at 95°C for 30 seconds, specific primer annealing at 59°C for 90 seconds, and extension at 72°C for 1 minute), ending with a final extension at 72°C for 10 minutes.
Arcobacter identification was performed by PCR amplification as described by Bastyns et al. [2] using ARCO1 (5′-GTCGTGCCAA GAAAAGCCA-3′) and ARCO2 (5′-TTCGCTTGCGCTGACAT-3′) primers, which amplified a 331-bp fragment of 23S rDNA.
All PCR reactions were performed with an automatic thermal cycler (Techne-Progene; Cambridge, UK). DNA templates from reference strains were used as positive controls. In addition, negative controls in which DNA was replaced with sterile distilled water were also included. All of the reagents used in the PCR reactions (Taq polymerase, dNTP, and MgCl2) were provided by Ecogen (Spain), and all of the primers employed were prepared by TIB MOLBIOL (Germany).
PCR products were detected by electrophoresis on 1.2% (wt/vol) agarose gel in 1 × Tris-acetate-EDTA (TAE) buffer at 100 V for approximately 60 minutes and visualized by staining with ethidium bromide (0.5 μg/mL) and ultraviolet (UV) transillumination. GeneRuler 100-bp DNA Ladder Plus (MBI Fermentas) was used as molecular marker.
PCR-RFLP analysis. A 1004-bp 16S rDNA fragment from all Helicobacter, Campylobacter, and Arcobacter strains was amplified using CAH16S 1a and CAH16S 1b primers with sequences 5′-AATA CATGCAAGTCGAACGA-3′ and 5′-TTAACCCAACATCTCACG AC-3′, respectively, according to the Marshall et al. assay [24]. For amplification of a 2.6-kbp fragment within the coding region of the 23S rRNA gene of Campylobacter, Arcobacter, and Helicobacter, primers LS1 (5′-GGATTTCCGAATGGGGCAACCC-3′) and LS2 (5′-GTTTCGTGCTTAGATGTTTC-3′) were used [15]. The PCR reaction was carried out as described by Hurtado and Owen [16], including an initial denaturation step at 95°C for 5 minutes and a final extension at 72°C for 5 minutes to ensure full extension of the product. In both cases, amplified products were visualised by 1% (wt/vol) agarose gel electrophoresis and stained with ethidium bromide (0.5 μg/mL).
PCR products (10 μl) were digested with 10 U restriction enzymes in a final volume of 20 μl at 37°C for 3.5 hours. Restriction endonucleases were selected by computer analysis using REbsites (available at: http://www.rebase.neb.com). DdeI (Roche) and HpaII (MBI Fermentas) were used to digest 16S rDNA and 23S rDNA fragments, respectively. The reaction was stopped by adding 3 μl stop-mix solution (50 mM EDTA, 0.3% Ficoll, and 0.3% bromophenol blue).
Restriction fragments were separated on 2% (wt/vol) agarose gel electrophoresis in TAE 1 × buffer at 90V for 3 hours and visualized after staining with ethidium bromide and UV transillumination. GeneRuler 100-bp DNA Ladder Plus was used as a standard for molecular size determination. To assess reproducibility for both 16S and 23S PCR-RFLP analysis, all of the strains were analysed at least two times in different experiments.
Results and Discussion
Biochemical identification
All of the clinical Helicobacter strains included in this study were Gram negative and positive for urease, catalase, and oxidase test. In previous genotypic analysis, 16 of the 20 Campylobacter strains were identified as C. jejuni and 2 as C. coli by the API-Campy system. The remaining two strains presented unacceptable profiles. Arcobacter strains were tested for growth at 30°C in aerobic conditions to confirm their aerotolerance, and after 24 to 48 hours of incubation, growth was observed in all of the plates. The API-Campy system was also applied, although all the isolates, including reference strains, were identified as A. cryaerophilus (Table 1). These results confirm the ineffectiveness of this system to identify Arcobacter strains. This fact has been previously reported by other investigators; Atabay et al. used the API-Campy scheme to study the diversity and prevalence of Arcobacter spp. in broiler chickens and found that only 20% of the isolates were correctly identified at the species level [1].
PCR identification
Helicobacter clinical and reference strains showed positive results by PCR assay using Helicobacter genus-specific primer sets C97 and C05. All of the clinical strains were identified as H. pylori by species-specific PCR. No amplification products were obtained from Arcobacter and Campylobacter reference strains.
A 491-bp fragment was obtained after amplification with genus specific primers only for Campylobacter spp. strains. After mPCR, 14 of the 20 Campylobacter strains were identified as C. jejuni and the remaining 6 as C. coli. For aerotolerant bacteria and Arcobacter reference strains, a 331-bp specific fragment was amplified with the primer set ARCO1/ARCO2 (Table 1).
PCR-RFLP differentiation
Bacterial strains previously identified as Helicobacter, Campylobacter, and Arcobacter were typed by PCR-RFLP of 16S rDNA and 23S rDNA technique (Table 1). The results of RFLP analysis resolved by agarose gel electrophoresis were almost identical to the predicted fragments based on the nucleotide sequence data.
A 1004-bp PCR product of the 16S rDNA was amplified in all of the strains. Differentiation to the genus level was achieved using DdeI because digestion of the amplicon with this restriction enzyme generated six different specific patterns: three for Helicobacter, two for Campylobacter, and one for Arcobacter (Fig. 1).

16S PCR-RFLP patterns of Helicobacter, Campylobacter, and Arcobacter reference strains generated by digestion with DdeI. Lane M = 100-bp DNA Ladder Plus with sizes indicated on right (bp). Lane 1 = H. pylori NCTC 11637. Lane 2 = H. pullorum NCTC 13153. Lane 3 = H. fennelliae NCTC 11612. Lane 4 = A. butzleri NCTC 12481. Lane 5 = A. cryaerophilus NCTC 11885. Lane 6 = A. skirrowii NCTC 12713. Lane 7 = C. jejuni NCTC 11828. Lane 8 = C. jejuni NCTC 12521. Lanes 9 and 10: C. coli NCTC 11366. Lane 11 = C. upsaliensis NCTC 11540.
Digestion with restriction enzyme DdeI yielded different specific patterns for H. pylori (495, 257, 102, and 75 bp); H. pullorum (750 and 230 bp); and H. fennelliae (750, 280, 170, and 140 bp). The 31 clinical isolates produced fingerprints that were identical to that of H. pylori reference strains.
Analysis of the 16S rRNA gene with DdeI enzyme was not useful for the specific identification of Campylobacter and Arcobacter. Identical fingerprints were obtained for all of the Arcobacter species tested (421, 353, and 183 bp). The method also failed to distinguish C. coli from C. jejuni. These two species shared the same DdeI profile (272, 247, 153, 120, and 95 bp). Only C. upsaliensis showed a species-specific pattern (425, 248, 145, and 108 bp). All of the Campylobacter isolates presented the same profile as C. jejuni and C. coli reference strains.
For 23S rDNA PCR-RFLP analysis, an internal region of the 23S rRNA gene of approximately 2.6-Kbp was amplified from all of the strains. The PCR products, showing a single band of the expected size, were subjected to restriction analysis with HpaII. The digestion produced different patterns that allowed for the discrimination among these three related genera (Fig. 2).

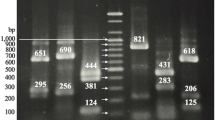
23S PCR-RFLP patterns of Helicobacter, Campylobacter, and Arcobacter reference strains generated by digestion with HpaII. Lane M = 100-bp DNA Ladder Plus with sizes indicated on right (bp). Lane 1 = H. fennelliae NCTC 11612. Lane 2 = H. pylori NCTC 11638. Lane 3 = H. pullorum NCTC 13153. Lanes 4 and 5 = C. coli NCTC 11366. Lane 6 = C. jejuni NCTC 11828. Lane 7 = C. jejuni NCTC 12521. Lane 8 = C. upsaliensis NCTC 11540. Lane 9 = A. cryaerophilus NCTC 11885. Lane 10 = A. skirrowii NCTC 12713. Lane 11 = A. butzleri NCTC 12481.
23S rDNA analysis of Helicobacter with HpaII enzyme also allowed for the identification of strains at the species level. Three different profiles were obtained, one for each one of the species studied: H. pylori (820, 600, 550, 320, and 230 bp); H. pullorum (600, 550, 480, 350, 305, and 230 bp); and H. fennelliae (110, 620, 320, 250, and 230 bp). Most of the clinical strains showed the same pattern (HpI) as H. pylori NCTC 11637 and NCTC 11638, but 3 of the 31 strains had a different profile (HpII: 1150, 600, 550, and 230 bp), which has not been described previously.
When applying the scheme to Campylobacter, this method allowed for the differentiation at the species level between C. jejuni and C. coli. Three different species-specific patterns were obtained: C. coli (682, 627, 558, and 456 bp); C. jejuni (627, 586, 558, 357, and 305 bp); and C. upsaliensis (1958 and 558 bp) profiles could be distinguished. Fourteen of the 20 Campylobacter strains were identified as C. jejuni and 6 as C. coli. The 2 strains that could not be identified by the API-Campy system were recognized by PCR-RFLP as C. jejuni and C. coli. In contrast, 3 strains characterized as C. jejuni by API-Campy presented the typical C. coli pattern, confirming the results previously obtained by mPCR. The API-Campy system seemed to have major problems in identifying C. coli strains because only 2 of the 6 such strains, on the basis of mPCR and 23S rDNA PCR-RFLP products, gave concordant results in API. Therefore, this system may not be reliable for the routine identification of Campylobacter species in accordance with other studies [17, 31]. A. butzleri showed a unique HpaII pattern (861, 615, 550, 320, and 240 bp), but A. skirrowii and A. cryaerophilus shared the same pattern (1107, 615, 550, and 240 bp). Seven of the 12 Arcobacter strains were identified as A. butzleri and the remaining 5 as A. skirrowii or A. cryaerophilus.
In all of the cases, the RFLP profiles were reproducible when a different batch of DNA was used in both PCR-RFLP analysis, and no variation in the restriction profiles of strains belonging to the same species was observed.
Our results show that the PCR-RFLP method is more reliable than biochemical identification for Helicobacter, Campylobacter, and Arcobacter. We agree with other investigators [3, 16, 24] that it is also faster and simpler than other molecular techniques that require large quantities of cells and involve complex and time-consuming steps. In addition, it is more profitable because it does not require numerous specific PCRs or expensive equipment.
In conclusion, PCR-RFLP analysis of 16S and 23S rRNA gene sequences allows for genus identification and achieves a good level of species differentiation with only one set of primers and one restriction enzyme. This technique is a fast, simple, and suitable alternative method to conventional identification procedures for reliable characterization of Helicobacter, Campylobacter, and Arcobacter species.
Literature Cited
Atabay HI, Corry JEL, On SLW (1998) Diversity and prevalence of Arcobacter spp. in broiler chickens. J Appl Microbiol 84:1007–1016
Bastyns K, Cartuyvels D, Chapelle S, et al. (1995) A variable 23S rDNA region is a useful discriminating target for genus-specific and species-specific PCR amplification in Arcobacter species. Syst Appl Microbiol 18:353–356
Cardarelli-Leite P, Blom K, Patton CM, et al. (1996) Rapid identification of Campylobacter species by restriction fragment length polymorphism analysis of a PCR-amplified fragment of the gene coding for 16S rRNA. J Clin Microbiol 34:62–67
Choi YK, Han JH, Joo HS (2001) Identification of novel Helicobacter species in pig stomachs by PCR and partial sequencing. J Clin Microbiol 39:3311–3315
Cover TL, Blaser MJ (1995) Helicobacter pylori: A bacterial cause of gastritis, peptic ulcer disease, and gastric cancer. ASM Features 61:21–26
de Boer WA, Tytgat GN (2000) Regular review: Treatment of Helicobacter pylori infection. Br Med J 320:31–34
Denis M, Soumet C, Revoal K, et al. (1999) Development of a m-PCR assay for simultaneous identification of Campylobacter jejuni and C. coli. Lett Appl Microbiol 29:406–410
Engvall EO, Brandstrom B, Gunnarsson A, et al. (2002) Validation of a polymerase chain reaction/restriction enzyme analysis method for species identification of thermophilic campylobacters isolated from domestic and wild animals. J Appl Microbiol 92:47–54
Fermér C, Engvall EO (1999) Specific PCR identification and differentiation of the thermophilic campylobacters, Campylobacter jejuni, C. coli, C. lari, and C. upsaliensis. J Clin Microbiol 37:3370–3373
Fox JG (2002) The non-H pylori helicobacters: Their expanding role in gastrointestinal and systemic diseases. Gut 50:273–283
Fox JG, Dewhirst FE, Shen Z, et al. (1998) Hepatic Helicobacter species identified in bile and gallbladder tissue from Chileans with chronic cholecystitis. Gastroenterol 114:755–763
Frost JA (2001) Current epidemiological issues in human campylobacteriosis. Symp Ser Soc Appl Microbiol 30:85S–95S
González I, Grant KA, Richardson PT, et al. (1997) Specific identification of the enteropathogens Campylobacter jejuni and Campylobacter coli by using a PCR test based on the ceuE gene encoding a putative virulence determinant. J Clin Microbiol 35:759–763
Hsueh PR, Teng LJ, Hung CC, et al. (1999) Septic shock due to Helicobacter fennelliae in a non-human immunodeficiency virus-infected heterosexual patient. J Clin Microbiol 37:2084–2086
Hurtado A, Owen RJ (1997a) A rapid identification scheme for Helicobacter pylori and other species of Helicobacter based on 23S rRNA gene polymorphisms. Syst Appl Microbiol 20:222–231
Hurtado A, Owen RJ (1997b) A molecular scheme based on 23S rRNA gene polymorphisms for rapid identification of Campylobacter and Arcobacter species. J Clin Microbiol 35:2401–2404
Huysmans MB, Turnidge JD, Williams JH (1995) Evaluation of API Campy in comparison with conventional methods for identification of thermophilic campylobacters. J Clin Microbiol 33:3345–3346
International Agency for Research on Cancer (1994) Schistosomotes, liver flukes and Helicobacter pylori. IARC Monogr Eval Carcinog Risks Hum 61:1–241
Jalava K, Hielm S, Hirvi U, Hanninen ML (1999) Evaluation of a molecular identification scheme based on 23S rRNA gene polymorphisms for differentiating canine and feline gastric Helicobacter spp. Lett Appl Microbiol 28:269–274
Jauk V, Neubauer C, Szolgyenyi W, Vasicek L (2003) Phenotypic and genotypic differentiation of Campylobacter spp. isolated from Austrian broiler farms: A comparison. Avian Pathol 32:33–37
Kabeya H, Kobayashi Y, Maruyama S, Mikami T (2003) One-step polymerase chain reaction-based typing of Arcobacter species. Int J Food Microbiol 81:163–168
Linton D, Lawson AJ, Owen RJ, Stanley J (1997) PCR detection, identification to species level, and fingerprinting of Campylobacter jejuni and Campylobacter coli direct from diarrheic samples. J Clin Microbiol 35:2568–2572
Mansfield LP, Forshythe SJ (2000) Arcobacter butzleri, A. skirrowii and A. cryaerophilus-potencial emerging human pathogens. Rev Med Microbiol 11:161–170
Marshall SM, Melito PL, Woodward DL, et al. (1999) Rapid identification of Campylobacter, Arcobacter, and Helicobacter isolates by PCR-restriction fragment length polymorphism analysis of the 16S rRNA gene. J Clin Microbiol 37:4158–4160
Melito PL, Woodward DL, Bernard KA, et al. (2000) Differentiation of clinical Helicobacter pullorum isolates from related Helicobacter and Campylobacter species. Helicobacter 5:142–147
Moreno Y, Ferrús MA, Vanoostende A, et al. (2002) Comparison of 23S polymerase chain reaction and amplified fragment length polymorphism techniques as typing systems for thermophilic campylobacters. FEMS Microbiol Lett 211:97–103
On SLW (2001) Taxonomy of Campylobacter, Arcobacter, Helicobacter and related bacteria: Current status, future prospects and immediate concerns. J Appl Microbiol 90:1S–15S
On SLW (1996) Identification methods for campylobacters, helicobacters and related organisms. Clin Microbiol Rev 9:405–422
O’Rourke JL, Grehan M, Lee A (2001) Non-pylori helicobacter species in humans. Gut 49:601–606
Pearson AD, Greenwood MH, Feltham RK, et al. (1996) Microbial ecology of Campylobacter jejuni in a United Kingdom chicken supply chain: Intermittent common source, vertical transmission, and amplification by flock propagation. Appl Environ Microbiol 62:4614–4620
Rautelin H, Jusufovic J, Hänninen ML (1999) Identification of hippurate-negative thermophilic campylobacters. Diagn Microbiol Infect Dis 35:9–12
Shen Z, Fox JG, Dewhirst FE, et al. (1997) Helicobacter rodentium sp. nov., a urease-negative Helicobacter species isolated from laboratory mice. Int J Syst Bacteriol 47:627–634
Solnick JV, Schauer DB (2001) Emergence of diverse Helicobacter species in the pathogenesis of gastric and enterohepatic diseases. Clin Microbiol Rev 14:59–97
Steinhauserova I, Ceskova J, Fojtikova K, Obrovska I (2001) Identification of thermophilic Campylobacter spp. by phenotypic and molecular methods. J Appl Microbiol 90:470–475
Stucki U, Frey J, Nicolet J, Burnens AP (1995) Identification of Campylobacter jejuni on the basis of a species-specific gene that encodes a membrane protein. J Clin Microbiol 33:855–859
Vandamme P, Falsen E, Rossau R, et al. (1991) Revision of Campylobacter, Helicobacter, and Wolinella taxonomy: Emendation of generic descriptions and proposal of Arcobacter gen. nov. Int J Syst Bacteriol 41:88–103
Weir SC, Gibert CL, Gordin FM, et al. (1999) An uncommon Helicobacter isolate from blood: Evidence of a group of Helicobacter spp. pathogenic in AIDS patients. J Clin Microbiol 37:2729–2733
Wesley IV (1997) Helicobacter and Arcobacter: Potential human foodborne pathogens? Trends Food Sci 89:293–299
Wilson K (1987) Preparation of genomic DNA from bacteria. In: Ausubel FM, Brent R, Kingston RE, Moore DD, Smith JA, Seidman JG, et al. (eds) Current protocols in molecular biology, unit 2.4.1. Wiley, New York, NY, pp 241–242
Acknowledgments
This work was supported by Research Project AGL2002-04480-C03-03 from Ministerio de Ciencia y Tecnología, Spain (National and FEDER fundings). A. González is the recipient of a Predoctoral FPU grant from Ministerio de Educación y Ciencia, Spain (AP2000-0795).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
González, A., Moreno, Y., González, R. et al. Development of a Simple and Rapid Method Based on Polymerase Chain Reaction–Based Restriction Fragment Length Polymorphism Analysis to Differentiate Helicobacter, Campylobacter, and Arcobacter Species. Curr Microbiol 53, 416–421 (2006). https://doi.org/10.1007/s00284-006-0168-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-006-0168-5