
Abstract
Beauveria brongniartii extracellular subtilisin-like serine protease (Pr1) is one of the most virulent factors by virtue of its activity against insect cuticles. The Pr1 cDNA was cloned using the switching mechanism at the 5′ end of the RNA transcript and rapid amplification of cDNA ends. The 1732-bp fragment of genomic DNA containing the predicted open-reading frame of the Pr1 gene was cloned by polymerase chain reaction and sequenced. The Pr1 cDNA is 1550 bp and contains an 1140-bp ORF. The deduced amino-acid sequence of the protein shows identity to that of proteinase K from Tritirachium album (62%), Pr1 from Metarhizium nisopliae (67%), and Pr1 from B. bassiana (76%). The Pr1 protein with an N-terminal fusion to the six-histidine tag was expressed in Escherichia coli as inclusion bodies with the expression vector pBV220. Sodium dodecylsulsulfate–polyacrylamide gel electrophoresis clearly revealed expressed product. The Pr1 protein was purified and refolded and had proteolytic activity of 0.288 U mg−1.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Entomopathogenic fungi can be used to control insects and have several advantages over chemical pesticides [14, 2]. They can actively invade hosts and have repeated infection cycles. They can survive in the soil during periods of low host densities, and the hosts themselves rarely acquire resistance to them. Entomopathogenic fungi produce extracellular, cuticle-degrading enzymes that include proteases, chitinases, and lipases [12]. These degrade the insect cuticle, which is an effective barrier against most microbial infections [5]. Extracellular, subtilisin-like serine protease Pr1 is the most important factor for the insecticidal activity of entomopathogenic fungi [1, 7]. Previously, the Pr1 genes from Metarhizium anisopliae and Beauveria bassiana were isolated and analyzed. The former gene was overexpressed in M. anisoplia, which greatly enhanced the strain virulence [14]. However, the Pr1 gene from B. brongniartii had not been cloned or sequenced. Because this fungus has a wide host range, we explored the mechanism of B. brongniartii infection at the molecular level.
In this article, we describe the cloning and analysis of the Pr1 cDNA and genomic DNA from B. brongniartii and the expression in Escherishia coli as part of our plan for improving B. brongniartii by genetic engineering.
Materials and Methods
Strains, plasmids, and culture conditions
The Pr1 gene was isolated from B. brongniartii 1019 (Institute of Agro-Environment and Sustainable Development, Chinese Academy of Agricultural Sciences, Beijing). E. coli DH5α (Life Technologies, Rockville, MD) was routinely used for transformation and propagation of plasmids. E. coli DH10B (Stratagene, La Jolla, CA) was used as the host strain for gene expression. pBV220 (Stratagene) was used to construct an expression vector. The pGEM-T vector (Promega, Madison, WI) was used as a cloning vector and also as a subcloning vector for DNA sequencing. B. brongniartii 1019 was cultured in SDY medium (40 g glucose l−1, 5 g peptone l−1, and 10 g yeast extract l−1), and expression of Pr1 was induced with protease induction medium (0.2 g K2HPO4 l−1 cicada exuviae extract and 0.2 g MgSO4·7H2O l−1) at 28°C. The cicada exuviae extract was prepared by boiling 10 g cicada exuviae (Chinese traditional medicine) l−1 for 30 minutes and passing it through filter paper. All E. coli strains were cultured in Luria Bertani medium [9].
Enzyme induction of the mycelia
After 60 hours of growth with shaking at 200 rpm, the mycelia of B. brongniartii 1019 were harvested and washed twice with sterile distilled water. Then the washed mycelia (2 mg ml−1) were transferred to the protease induction medium and cultivated with shaking at 200 rpm. The supernatant was collected as crude enzyme after centrifugation for 10 minutes at 8000 g [3].
Enzyme assay
The enzyme activity of Pr1 was measured according to St. Leger et al. [11]. Suc-Ala-Ala-Pro-Phe-pNA (Sigma) in 1 mM DMSO was used as substrate. One unit of Pr1 activity is defined as an increase of 0.1 unit A410 per minutes. The protein concentration was measured using the Coomassie Blue G-250 binding assay with BSA as the standard.
Extraction of total DNA and RNA from B. brongniartii
Genomic DNA was isolated using the method of St. Leger et al. [13] from 3-day cultures of B. brongniartii that were grown in liquid SDY medium. Total RNA was extracted from finely comminuted frozen mycelia of B. brongniartii using a UNIQ-10 Trizol RNA Extraction Kit (Sangon, Shanghai, China).
Cloning an internal of the Pr1 cDNA
Reverse transcription–polymerase chain reaction (RT-PCR) was performed carried out using the Access RT-PCR System (Promega). Based on the highly conserved regions of several known subtilisin-like serine protease genes [7], a pair of primers, P1-F [5′-TGGGGTCTAGGTCGCA TCTC-3′] and P1-R [5′-GCCAGGTGCGAAAATGTCAAC-3′], were designed. A 654-bp DNA fragment was amplified by RT-PCR from 10 μg total RNA.
5′-RACE PCR and 3′-RACE PCR
Unlike cloning the Pr1 cDNAs from M. anisopliae [14] and B. bassiana [7], the full-length Pr1 cDNA of B. brongniartii was obtained by SMART-RACE. Two pairs of gene-specific primers were designed for 5′-RACE PCR and 3′-RACE PCR based on the acquired internal fragment sequence. For 5′-RACE PCR, the external primer was GSP1 [5′-GAGGTGTTGGCGGCATCACGA TTG-3′], and the internal one was NGSP1 [5′-CGACAGCGACAA AAACGCCCGAAG-3′]. For 3′-RACE PCR, the external primer was GSP2 [5′-GGACCGGAGAACACGAAGCGAATG-3′], and the inter- nal one was NGSP2 [5′-CGCCATCGCCAGCATGTCCCTTG-3′]. The internal primers were used to increase the specificity. The location of the primers and the cDNA template is shown in (Fig. 1a). PCR was performed at 94°C for 1 minute followed by 5 cycles at 94°C for 5 seconds and at 72°C for 3 minutes. This was followed by 5 cycles at 94°C for 5 seconds, at 70°C for 10 seconds, and at 72°C for 3 minutes and by 40 cycles at 94°C for 5 seconds, at 68°C for 10 seconds, at 72°C for 3 minutes, and at 72°C for 7 minutes. The 5′ end (929 bp) and the 3′ end (793 bp) RACE products of the Pr1 cDNA were amplified by 5′-RACE PCR and 3′-RACE, respectively, and then sequenced.

(a) The location of gene-specific primers in the cDNA template. (b) The structure of B. Brongniartii Pr1 gene.
Amplification of full-length Pr1 cDNA and genomic DNA
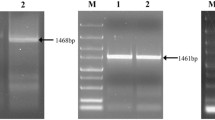
The upstream primer, P2-F [5′-GCGGGGAAGCAAGCAAGCAAG-3′], was designed based on the sequence of the 5′-RACE product, whereas the downstream primer, P2-R [5′-ATTGAATCGGTGGTAA ATAGCCTC-3′] was designed based on the sequence of the 3′-RACE product. The pair of primers was used to clone the full-length cDNA and genomic DNA encoding Pr1. The full-length cDNA (approximately 1500 bp) and genomic DNA (approximately 1700 bp) encoding Pr1 were amplified using total cDNA and genomic DNA, respectively, as templates.
DNA sequencing and analysis
The PCR amplification products were cloned into the pGEM-T vector (Promega) and sequenced by Shanghai Bioasia Engineering (China). The DNASIS and DNASTAR programs were employed to analyze the DNA sequences.
Plasmid construction
Pr1-6His contains the complete open reading frame (ORF) from the Pr1 cDNA that encodes the Pr1 protein plus the sequence encoding six histidines on its N-terminus. This sequence was amplified with the 5′ primer P3-F [5′-CAGCGAATTCATGCATCA TCACCATCATCACCGTCTATCAATCATCGCAGCC-3′], with an added EcoRI restriction site (bold), and the sequence encoding the 6 His tag and the 3′ primer P3-R [5′-GCCGGAATTCACTGGCTGAA AAACACGTGCT-3′], also with an added EcoRI restriction site (bold). The amplified fragments were digested with EcoRI and inserted into the EcoRI site of pBV220, producing the expression plasmid pBV220-Pr1-6His (Fig. 2).

The construction of expression plasmid pBV220-Pr1-6His. The Pr1 gene is under the control of PRPL tandem of λ bacteriophage for the high levels of expression.
Enzyme induction in E. coli
E. coli transformants were cultured overnight. The temperature was increased to 42°C, and the cultures were incubated for another 4 hours to induce the expression of target proteins. The cells were harvested by centrifugation at 4000 rpm for 20 minutes. The expression level and the localization (supernatant or insoluble fraction) of the objective proteins were analyzed by gel-analysis software after SDS-PAGE.
Separation of inclusion bodies
Induced E. coli cells were centrifuged at 4000 g for 30 minutes, and the cell pellet was dissolved in 50 mM Tris-HCl buffer (pH 8.0) containing 5 mM ethylene diamine tetraacetic acid and 1 mM PMSF. Cells were lysed by sonication and centrifuged at 8000 g for 30 minutes to isolate the Pr1-6His inclusion bodies. Then the inclusion bodies were recovered by centrifugation at 8000 g. The inclusion body pellets were washed with 50 mM Tris-HCl buffer (pH 8.0) containing 5 mM EDTA and 2% deoxycholate [8]. Finally, the inclusion bodies were washed with distilled water to remove contaminating salt and detergent and centrifuged at 8000 g for 30 minutes. At this stage, the inclusion bodies contained mostly Pr1-6His.
Solubilization and purification of Pr1-6His from inclusion bodies
Different buffers with various pH levels—and containing different denaturants, salts, ionic detergents, or oxidizing and decreasing agents—were tested for solubilization of the Pr1-6His inclusion bodies. Finally, the purified Pr1-6His inclusion bodies were solubilized in 100 mM Tris buffer (pH 8) containing 8 M urea, 150 mM NaCl, 1 mM EDTA, and 0.5% β-mercaptoethanol for 60 minutes at room temperature. The solution resultant was centrifuged at 12,000 rpm for 30 minutes, and the supernatant was collected. This supernatant was applied to an Ni2+-chelating Sepharose column (25 ml) (Qiagen, Santa Clarita, CA). The column was equilibrated with buffer (10 mM Tris-HCl and 30 mM imidazole), and the sample was loaded at a concentration of 1.5 mg/ml. The column was washed with 100 ml equilibration buffer, and proteins were eluted using elution buffer (10 mM Tris-HCl and 500 mM imidazole) [6]. Fractions, 4 ml, were collected, and portions of the fractions were analyzed by SDS-PAGE.
Refolding the fusion bodies
The Pr1-6His was then refolded during four dialysis steps with 50 mM Tris-HCl buffer (pH 8.0), 1 mM EDTA, and 0.1% β-mercaptoethanol containing decreasing concentrations of imidazole (400, 200, 50, and 0 M) at 4°C. The precipitate was removed by centrifugation at 12,000 rpm for 20 minutes, and the supernatant was collected.
Results and Discussion
Activity of B. brongniartii Pr1 in the protease induction medium
Because Pr1 is induced when insect cuticles are the sole carbon and nitrogen source, B. brongniartii mycelia were transferred from liquid SDY medium to the protease induction medium. The Pr1 activity and the dry weight of the mycelia were measured during cultivation times (Fig 3). The results indicated that the dry weight of the mycelia did not increase in the protease induction medium. The peak of Pr1 specific activity is shown in (Fig. 3). The peak of Pr1 specific activity was at 50 hours, and therefore the mycelia were harvested at 40 hours and 50 hours and mixed for total RNA extraction.

Time course of the specific activity of Pr1 and the dry weight of B. Brongniartii mycelia in the protease induction medium. Triangles = mycelial dry weight; squares = Pr1-specific activity
To clone the full-length Pr1 cDNA, a 654-bp internal fragment of the Pr1 cDNA was first amplified by RT-PCR from B. briongniartii and sequenced. The sequence was analyzed with BLAST programs. Analysis demonstrated that the 654-bp fragment is a part of the Pr1 cDNA. The full-length cDNA (approximately 1500 bp) and genomic DNA (approximately 1700 bp) were amplified.
The genomic DNA encoding Pr1 in B. brongniartii is 1732-bp long and contains three introns that are 64-, 57-, and 61-bp long, respectively (Fig. 1b). All of the introns have the boundary sequences 5′ (exon)/GT... AG/(exon) 3′, which is characteristic of most of eukaryotic genes. Except for the poly(A) tail, the cloned cDNA is 1550-bp long and contains an ORF of 1140 bp together with 342-bp 5′ and 68-bp 3′ untranslated regions. The ORF encodes 380 amino-acids residues, including a typical 18 amino-acid hydrophobic N-terminal signal peptide. The calculated molecular mass of the Pr1 precursor is 39121 Da, and its pI is 8.0. The protein also has three active sites (D140, H170, and S326), which are characteristic of the subtilisin-like serine proteases. The GenBank accession nos. for the Pr1 cDNA and genomic DNA sequence are AY520815 and AY520814, respectively.
The predicted amino-acid sequence of B. brongniartii Pr1 has a high homology to other members of the subtilisin family of serine proteases (Fig. 4). The predicted amino-acid sequence of Pr1 showed 62%, 67%, and 76% identity to that of Tritirachium album proteinase K (ProK) (P06873) [4], M. anisopliae Pr1 (MaPr1) (M73795) [13], and B. bassiana Pr1 (BbPr1) (U16305) [7], respectively. As expected, the active sites (D140, H170, and S326) of Pr1 in B. brongniartii were highly conserved. (Fig. 4) shows that the nonhomologous sequences among the Pr1 proteins were mainly at the C termini. This is probably the reason that we previously failed to obtain the Pr1 gene from B. brongniartii with primers based on the 5′and 3′ ends of Pr1 gene [4, 7] from the most closely related species, B. bassiana.

Alignment of the deduced B. Brongniartii Pr1 amino-acid sequence with subtilisin family enzymes, T. album proteinase K (ProK), M. anisopliae Pr1 (MaPr1), and B. bassiana (BbPr1). Gray boxes = identical amino acids between B. Brongniartii Pr1 and other proteases.
Construction of a recombinant E. coli strain and its Pr1 expression
Plasmid pBV220, containing the PRPL tandem promoters of λ bacteriophage for high levels of expression and the cIts857 restraining gene of λ bacteriophage adapted for heat-induced expression, was used for Pr1-6His expression in E. coli. DNA fragments with the complete ORF, including the signal peptide from the Pr1 cDNA template and the sequence encoding the 6His tag, were inserted into pBV220, resulting in pBV220-Pr1-6His. pBV220-Pr1-6His was transferred into E. coli DH10B. The transformant was designated DHPr1. After induction at 42°C for 4 hours, SDS-PAGE image analysis showed that the DHPr1 cells produced a large amount of a new protein (molecular weight approximately 40 kDa) in the form of inclusion bodies (Fig. 5). The fusion protein Pr1-6His was renaturated by dialysis and purified on a Ni-NTA column (Fig. 5). The Pr1 proteolytic activities were tested after dialysis (Table 1.) Significant proteolytic activity was detected compared with the control strain.

Samples from Pr1-6His purification steps. Proteins were separated by SDS-PAGE (11%) and visualized by Coomassie brilliant blue G-250 staining. Lane M = molecular mass standards; lane 1 = lysate from E. coli DHN cells 4 hours after induction; lane 2 = lysate from E. coli DHPr1 cells 4 hours after induction; lane 3 = protein Pr1-6His after Ni2+-NTA and after refolding.
Literature Cited
Charnley AK, St. Leger RJ (1991) The role of cuticle–degrading enzymes in fungal pathogenesis in insects. In: Cole ET, Hoch HC (eds) Fungal spore and disease initiation in plants and animals. New York, NY: Plenum, pp 267–287
Clarkson JM, Keith CA (1996) New insight into the mechanisms of fungal pathogenesis in insects. Trends Microbiol 5:197–203
Fang WG, Zhong YJ, Fei Y (2002) Cloning and characterization of cuticle degrading enzyme CDEP-1 from Beauveria bassiana. Chin J Genet 29:278–282
Gunkel FA, Gassen HG (1989) Proteinase K from Tritirachium album Limer: Characterization of the chromosomal gene and expression of the cDNA in Escherichia coli. Eur J Biochem 179:185–194
Kim HK, Hoe HS, Suh DS, Kang SC, Wang CH, Kwon ST (1999) Gene structure and expression of gene from Beauveria bassiana encoding bassiasin I, an insect cuticle-degrading serine protease. Biotechnol Lett 21:777–783
Hainfeld JF, Liu W, Halsey CM, Freimuth P, Powell RD (1999) Ni-NTA-gold clusters target His-tagged proteins. J Struct Biol 127:185–198
Joshi L, St. Leger RJ, Bidochka MJ (1995) Cloning of a cuticle-degrading protease from the entomopathogenic fungus, Beauveria bassiana. FEMS Microbiol Lett 125:211–218
Khan RH, AppaRao KBC, Eshwari ANS, Totey SM, Panda AK (1998) Solubilization of recombinant ovine growth hormone with retention of native like secondary structure and its refolding from the inclusion bodies of E. coli. Biotechnol Prog 14:722–728
Sambrook J, Fritsch EF, Maninatis T (1989) Molecular cloning: a laboratory manual, 2nd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press
St. Leger RJ (1995) The role of cuticle-degrading proteases in fungal pathogenesis of insects. Can J Bot 73:1119–1125
St. Leger RJ, Charnley AK, Cooper RM (1987) Characterization of cuticle degrading protease produced by the entomopathogen Metarhizium anisopiae. Arch Biochem Biophys 253:221–232
St. Leger RJ, Cooper RM, Charnley AK (1987) Production of cuticle-degrading enzymes by the entomopathogen Meterhizium anisopliae during infection of cuticles from Calliphora vomitoria and Manduca sexta. J Gen Microbiol 133:1371–1382
St. Leger RJ, Frank DC, Roberts DW, Staples RC (1992) Molecular cloning and regulatory analysis of the cuticle-degrading-protease structural gene from the entomopathogenic fungus, Metarhizium anisoplia. Eur J Biochem 204:991–1001
St. Leger RJ, Joshi L, Bidochka MJ, Roberts DW. (1996) Construction of an improved mycoinsecticide overexpressing a toxic protease. Proc Natl Acad Sci U S A 93:6349–6354
Acknowledgments
This work was supported by the National Doctoral Foundation of China (Grant No. 20040422056); the Opening Fund of the State Key Laboratory of Microbial Technology, Shandong University, China; and the National High Technology Research and Development Program of China (Grant No. 863-2003AA241130). In addition, we are grateful to Dr. Greenwood for manuscript editing.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sheng, J., An, K., Deng, C. et al. Cloning a Cuticle-Degrading Serine Protease Gene with Biologic Control Function from Beauveria brongniartii and Its Expression in Escherichia coli. Curr Microbiol 53, 124–128 (2006). https://doi.org/10.1007/s00284-005-5336-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-005-5336-5