
Abstract
A gene encoding an extracellular protease from Dichelobacter nodosus was characterized and expressed in E. coli rosetta-gami (DE3). The nucleotide sequence analysis revealed an ORF of 1427 bp ecoding 475 amino acids long protein of calculated molecular weight 50.6 kDa and pI value 6.09. The phylogenetic analysis showed relatedness to subtilisin-like serine proteases of peptidase S8 family. The amino acid sequence analysis showed presence of N-terminal pre-peptide (1–23 aa), pro-peptide (24–160 aa), peptidase S8 domain (161–457 aa), and a C-terminal extension (458–475 aa). The gene harboring native signal peptide was expressed in pET-22b(+) for production of AprV2 recombinant protein. SDS-PAGE revealed the highest production of IPTG induced recombinant protein ∼37 kDa at 16 °C after 16 h. The purified protein after Ni-NTA affinity chromatography showed single protein band of ∼37 kDa which was also confirmed by the detection of blue coloured band of same size in Western blotting. The recombinant protein showed activity over broad temperature and pH range with optimum at 35 °C and pH 7.0. Similarly, the enzyme was stable over broad range 15–65 °C and 4–10 pH with maximum stability at 25 °C and pH 6. The activity of purified enzyme was also stimulated in the presence of Ca2+. The purified enzyme showed highest activity towards casein as compared to gelatin and BSA. These findings suggest AprV2 as an important candidate for industrial applications such as pharmaceuticals.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dichelobacter nodosus is an anaerobic, Gram-negative bacterium responsible for causing footrot disease in ruminants [1, 2]. Footrot is characterized by redness and soreness of the hoof skin leading to extreme lameness and failure of body functions [3, 4]. The proteases secreted by D. nodosus are important virulence factors enabling bacteria to establish in hoof of the affected animal. They act by digesting the tissue of the hooves, colonize themselves, and initiate the pathogenesis.
The proteases are produced as inactive precursors in the cytoplasm and converted to active mature enzyme in the extracellular environment after autocatalytic processing [5]. Several precursor proteases have been identified from pathogenic bacteria Bacillus anthracis, Serratia sp., Streptococcus pneumoniae, Vibrio sp., and Yersinia pestis [6–8]. Proteases maintain the structure of various organs and functions like blood clotting, the lysis of blood clots, processing and transport of secretory proteins across membranes and as pathogenic factors by inducing restricted proteolysis [9]. Proteases are also involved in the treatment of heart and lung disorders, digestive and eye disorders, soreness and ulcers in skin [10]. Accordingly, proteases are gaining attention in medical research and drug development as little information is available on their therapeutic applications [11, 12].
In the present study, AprV2 gene has been cloned and expressed in heterologous host and purified recombinant protein has been characterized for its molecular weight, optimum temperature and pH, metal ions and substrate specificity with suitability for use as therapeutic agent.
Materials and Methods
Isolation and Identification of D. nodosus
D. nodosus previously isolated from a case of virulent footrot from goat was revived by adding 1 ml of tripticase arginine serine (TAS) broth to the lyophilized culture [13] and streaked on TAS agar with 2 % hoof powder. The plate was incubated in an anaerobic jar at 37 °C for 4 days. Morphologically suspected colonies were confirmed using Gram staining.
Molecular Characterization of D. nodosus
The genomic DNA was isolated from pure bacterial culture and amplification of 16S rRNA gene corresponding to variable regions of D. nodosus was carried out using PCR [14]. PCR reaction was carried out in 50 μl reaction volume composed of 2 μl (50–100 ng) of template DNA, 1 μl of each 10 μM primer, 0.5 μl of 10 mM dNTPs mix, 5 μl of 10× Taq buffer and 0.25 μl (5 U μl−1) of Taq polymerase (Promega Corporation, USA). The final volume was adjusted using nuclease-free water. PCR amplification was performed in thermal cycler (Applied Biosystems, USA) at 94 °C for 2 min followed by 5 amplification cycles of 94 °C for 30 s, 62 °C for 30 s, and 72 °C for 30 s, and 25 amplification cycles involving denaturation at 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 30 s, with a final extension at 72 °C for 8 min. The amplicons were separated on 1 % (w/v) agarose gel stained with ethidium bromide. Serogrouping of D. nodosus was done based on fimbrial gene (fimA) using primers specific to A-I serogroup [15]. Virulence testing of the isolate was carried out by the detection of IntA gene [16] and gelatin gel protease thermostability assay [17].
Cloning and Sequencing of Protease Gene (AprV2)
The genomic DNA was used for the amplification of gene encoding the entire coding region and 5′ and 3′ flanking regions of complete AprV2 protease. The complete ORF encoding a precursor protease along with signal peptide sequence was amplified using primer pairs with inbuilt NdeI/Xho1 restriction sites 5′-GAC AGA ATT CCC ATA TGA AAC GAT TCA TTA TGA ACA AAA TG-3′ and 5′-TGT AAG CTT CTC GAG ACG ATT GCC TTC-3′. Amplification was carried out using Pfu DNA Polymerase (Promega Corporation, Madison, USA), and the purified PCR product was ligated into pET-22(b)+ plasmid pre-digested with the same restriction enzymes and transformed into E. coli DH5α, and the construct was named as pET-AprV2. Positive clones were confirmed by colony-PCR and final confirmation was done using nucleotide sequencing from First BASE laboratories Sdn, Bhd, Selangor, Malaysia. The sequence similarity search was done using BLASTN of National Center for Biotechnology Information [18]. The amino acid sequence was obtained from nucleotide sequence using Expasy translate tool (http://www.expasy.org/) and domains were predicted using Simple Modular Architecture Research Tool (SMART) [19]. The catalytic triad, active site residues, and conserved domains were analyzed using NCBI-CDD (www.ncbi.nlm.nih.gov/cdd) [20]. The protein sequences were retrieved from the MEROPS and NCBI database and phylogenetic tree was prepared in Mega 5 using neighbor-joining method [21].
Structural Features
Based on the crystal structure of peptidase domain of acidic extracellular subtilisin-like protease AprV2 from D. nodosus VCS1703A (PDB ID-3PLA; Uniprot ID A5EXI3), the structure was modeled by automated modeling using SWISS-MODEL (http://swissmodel.expasy.org) [22]. The quality of final model was assessed with QMEAN server [23]. PROCHECK was performed to assess the stereo-chemical qualities of the 3D models. The model was validated using Structural Analysis and Verification Server version 4.
Expression and Purification of Recombinant Protease
For expression analysis, the recombinant plasmid was further transformed into E. coli rosetta-gami (DE3) with 50 μg ml−1 ampicillin and 34 μg ml−1 chloramphenicol. The clones harboring recombinant plasmid grown in LB medium were induced with 1 mM IPTG at 16 °C. For time course expression analysis, samples were harvested after 4, 8, and 16 h for optimum production of recombinant protein. The harvested cells were resuspended in 50 mM Na2HPO4, 300 mM NaCl, pH 8.0 buffer for lysis and disrupted by sonication and centrifuged to collect the supernatants. The fractions were analyzed on SDS-PAGE for expression of the recombinant protein, and molecular weight was determined using unstained protein molecular weight marker (Invitrogen) after staining with Coomassie Brilliant Blue R-250.
The recombinant protein was purified using His-Trap Ni-NTA column (GE Healthcare, Sweden) and analyzed on SDS-PAGE after staining with CBBR-250. The detection of recombinant protein was confirmed on Hybond-P PVDF membrane using primary 6x-His tag monoclonal mouse antibodies HRP-conjugate in the presence of chromogenic substrate 4-chloro-1-naphthol for 1 h in Western blotting.
Protease Assay
Protease activity was measured by modifications in Folin and Ciocalteu’s reagent-based assay [24, 25]. The reaction was carried out using 25 μl of purified protease mixed with 130 μl of 0.6 % casein. The mixture was incubated at 37 °C for 30 min and stopped after addition of 130 μl of 10 % trichloroacetic acid. The reaction mixture was centrifuged at 12,000 rpm for 5 min and short peptides and free amino acids were determined by adding Folin-Ciocalteu’s phenol reagent (Sigma-Aldrich, USA). One unit of enzyme activity determines the enzyme that liberated 1 μmole of tyrosine per minute. Standard curve was prepared using known concentrations of tyrosine.
Characterization of Recombinant Protease
The optimum temperature for protease activity was determined at a temperature range of 15–65 °C for 30 min. The stability was measured by keeping the enzyme at different temperatures for 1 h before adding the substrate, and activity was measured under standard conditions. The pH profile of protease activity was determined at 4–10 pH using 100 mM sodium acetate (pH 4–5), 100 mM sodium phosphate buffer (pH 6–7), 100 mM Tris–HCl (pH 8–9), and 100 mM Glycine-NaOH buffer (pH 10) [25]. Similarly, the pH stability was determined by incubating the enzyme in buffers of different pH (4–10) for 1 h and enzyme activity was measured under standard assay conditions.
The effect of metal ions Ca2+, Mg2+, Na+, Cu2+, and EDTA at a concentration of 10 mM was studied by pre-incubating the metal ions with purified enzyme for 1 h [26]. The residual activity was measured by Folin-Ciocalteu’s phenol reagent as described above. The activity measured without metal ions was considered as 100 % enzyme activity.
The specificity of the purified enzyme for different substrates was also tested with casein, BSA, and gelatin. The protease activity was measured under standard assay conditions, and casein was taken as control.
Statistical Analysis
The enzyme assays were done in triplicates. Differences between mean values were determined, and standard error was indicated by vertical bars in figures. ANOVA was performed, and mean values were compared by CD value at p = 0.01 in tables. All the statistical analyses were performed using STATISTICA version 10.
Nucleotide Sequence Accession
The nucleotide sequence was submitted to NCBI-GenBank with accession number JQ082891.
Results and Discussion
Identification of D. nodosus
The bacterial strain D. nodosus appeared as transparent colonies with elevated centers on TAS agar after 4 days and curved Gram-negative rods with bulged ends in staining. The strain identified based on 16S rRNA gene showed PCR product of ∼783 bp.
The serotyping based on fimA and intA gene classified bacterial isolate as serotype B and a virulent strain showing amplified PCR products of 283 and 530 bp, respectively, which is also supported by the liquefaction of gelatin in well plate assay.
Cloning and Sequencing of the Protease Gene
The AprV2 gene cloned in E. coli-rosetta gami using pET-22b(+) vector gave sequence length of 1760 bp containing the entire coding region and 5′ and 3′ flanking regions in comparison to 1422 bp (FN674446) [27], 1803 bp (ABQ13853) [28], and 2160 bp (L38395) [29] reported in D. nodosus. The deduced amino acid sequence displayed 99.8, 99.6, and 97 % homology with peptidase S8, V2 protease, and acidic protease V2 from D. nodosus, respectively (Table 1). The phylogenetic analysis of AprV2 showed close relatedness and clustering with the acidic serine proteases belonging to the peptidase S8 family (Fig. 1). The homology based on amino acid sequences of S8 family showed clustering of AprV2 serine protease with various subtilisns of bacterial origin in S8A subfamily (Fig. 2).

Phylogenetic analysis of AprV2 protease from Dichelobacter nodosus based on amino acid sequence representing different clusters by using neighbor-joining method. Values shown next to branches are the percentage replicate tree with associated proteases clustered in the bootstrap test. The evolutionary analysis was conducted in MEGA 5 and distances computed using Kimura 2-parameter method. GenBank accession numbers of proteases are also provided

Neighbor-joining tree of AprV2 with members of the S8A subfamily of serine proteases based on amino acid sequences. Bootstrap values are expressed as percentages of 1000 replications and are shown at the nodes. The evolutionary analysis was conducted in MEGA 5 and distances computed using Kimura 2-parameter method
Sequence Analysis and Structural Features
The sequence analysis of AprV2 revealed an ORF of 1427 nucleotides encoding 475 amino acids long protein with calculated molecular weight 50.6 kDa and theoretical pI value 6.09. The analysis of AprV2amino acid sequence showed a conserved catalytic domain of peptidase S8 family along with the presequence and prosequence in the protein. The signal peptide of 23 amino acids with cleavage site between Ser23 and Ala24 residues followed by a propeptide between 24 and 160 amino acid residues, a mature peptidase domain between 161 and 457 amino acid residues, and small C-terminal extension of 458–475 amino acids (Fig. 1S, supplementary data). AprV2 is secreted as a precursor protein similar to the precursor proteases reported in Bacillus subtilis, Bacillus alcalophilus, Bacillus licheniformis, D. nodosus, and Staphylococcus aureus [30–33]. Three residues Asp169, His233, and Ser405 located in the mature domain of AprV2 formed a catalytic triad center (Fig. 1S), which is a characteristic feature of subtilisin-like serine proteases [34]. The geometric orientations of these residues are common but arrangement order of these amino acids in trypsin-like domain is HDS and SDH in carboxypeptidases [35].
A comparison of the predicted 3D structure of AprV2 with template 3PLA_A predicted 21.5 % helices, 20.9 % sheets, and 57.6 % loops (Fig. 3a) smoothly superimposed on the template. The mature domain of AprV2 protease modeled using template 3PLA_A with 100 % sequence identity and 61 % sequence similarity gave QMEAN score 0.733 within the prescribed limits (0–1) represented good quality and closeness of the model with the experimentally validated structure of 3PLA_A. Ramachandran plot depicted 85.1 % amino acid residues in the most favored regions, 14.6 % amino acid residues in the additional allowed regions, and only 0.3 % amino acid residues in the disallowed conformations. The alignment of model protein with template 3PLA_A showed root mean squares deviation (RMSD) of 0.1 Å indicating the complete superimposition of atoms on the template and showed validation score of 99.7 % in VERIFY_3D, reflecting good quality of the protein model. AprV2 also showed the presence of conserved active site residues D169, H233, V239, S305, L306, G307, A333, G335, N336, and S405 in the peptidase domain which appeared closely in the 3D structure forming a pocket but distantly placed in the primary structure (Fig. 3b).

a Structural analysis of AprV2 protease from Dichelobacter nodosus based on the available 3D structure of 3PLA in PDB showing helices, sheets, and catalytic triad D169, H233, and S405 in colored spheres. b Surface diagram of AprV2 protease showing active site residues in different colors against the gray background. The graphical presentations were made in PYMOL
Purification and Characterization of the Recombinant Enzyme
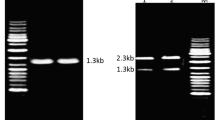
SDS-PAGE showed highest production of recombinant protein at 16 °C after 16 h induction with 1 mM IPTG (Fig. 4a). However, in similar studies, maximum recombinant protease production has been reported from 16–30 °C after 16-h induction in E. coli from D. nodosus [33, 36]. The recombinant proteins purified using Ni-NTA affinity showed single protein band of ∼37 kDa (Fig. 4b) which was also confirmed by the detection of blue colored band of the same size in Western blot analysis (Fig. 4c). These results indicated the removal of pre-propeptide region ∼20 kDa from the precursor protein as the ∼37 kDa mature protein along with His-tag was detected on SDS-PAGE. Similarly, the purified protein of size ∼37 kDa has already been reported in various strains of D. nodosus [33]. The processing of precursor protease AprV2 appeared responsible for variation in the size of mature protein which was predicted 50.6 kDa at nucleotide level and observed ∼37 kDa at the protein level in SDS-PAGE. These cleavable propeptides in the precursor proteins act as intramolecular chaperone to maintain protein conformation but are not involved in the translocation of protein [37]. Thus, it was indicated that C and N-terminal processing in AprV2 is important for the secretion of mature active protease to the extracellular medium. The uncleaved prosequence inhibits activity on interaction with the active site of catalytic domain [38].

a Time course analysis of recombinant protein AprV2 expressed in E. coli rosetta-gami (DE3): 1, uninduced protein after 4 h; 2, IPTG induced protein after 4 h; 3, uninduced protein after 8 h; 4, IPTG induced protein after 8 h; 5, uninduced protein after 16 h; 6, IPTG induced protein after 16 h. b Purification of 6x-His tagged recombinant protein using affinity chromatography: 1, IPTG induced recombinant protein AprV2; 2, purified protein after Ni-NTA chromatography. c Confirmation of 6x-His tagged recombinant proteins using Western blot assay: 1, IPTG induced recombinant protein AprV2 after chromatography; M, prestained protein molecular weight marker
The protease activity of AprV2 after cleavage of pre-propeptides was confirmed using casein, showing 98.4 U mg−1 increase in specific activity after purification. The purified protease was active over temperature range 15 to 65 °C and pH 4–10 with optimum activity at 35 °C and pH 7 (Fig. 5). In similar studies, the recombinant proteases have been reported optimally active at 25–65 °C and pH 7–8 from Bacillus spp., Colwellia sp., D. nodosus, and Pseudoalteromonas spp. [30, 39–43]. Similarly, the purified protease was stable over temperature 15–55 °C and pH 5–10 (Fig. 5). The serine proteases have also been reported stable at 35–50 °C from goat skin surface metagenome and metagenomic cosmid library from the coastal sediments near Antarctic [44, 45]. The effect of metal ions showed significant increase in protease activity by Ca2+ and retained more than 90 % activity in the presence of Na+, Mg2+, and Cu2+ as shown in supplementary data (Table 1S), similar to the effect of metal ions on proteases from Flavobacterium psychrophilum, Flavobacterium balustinum, and Serratia rubidaea [46–48]. Ca2+ has been reported to maintain stable conformation and protection against thermal denaturation of protein. Mg2+ and Cu2+did not show much effect on the activity while retained 57 % activity in the presence of EDTA (Table 1S) similar to the proteases reported from Colwellia sp. [40], B. subtilis [42], and Serratia marcescens [49]. The purified protease showed 100 % activity for casein, 95 % for BSA, and 68 % for gelatin given as supplementary information (Table 2S), which is an important feature of proteases to discriminate among competing substrates; also advantageous for use in pharmaceutical applications [50, 51]. These reported proteases have role in industrial applications like detergent, leather and food processing. But, there is no report on functional characterization of AprV2 protease from D. nodosus from India. The properties like stability and activity over broad range of temperature and pH, and substrate selectivity also suggested its potential and suitability for use in therapeutic applications like skin debridement, anti-inflammatory, antimicrobial, and clot dissolving agents. There is an immense need to explore such bacteria competent in producing proteases of commercial potential.

Effect of temperature a and pH b on activity and stability of mature protease AprV2 of D. nodosus expressed in E. coli rosetta-gami (DE3). The activity was measured using casein as substrate. One unit of activity corresponds to 1 μmol of the reaction product released from substrate for 1 min at optimal temperature and pH. The values are the mean of three experimental repeats with SE
Conclusions
The recombinant protease was found subtilisin-like acidic serine type of peptidase S8 family. The protease expressed in E. coli rosetta-gami (DE3) using pET-22b(+) vector encoded ∼50.6 kDa protein, resulted in ∼37 kDa mature protein after removal of signal peptide and pre-propeptide region. The processing of precursor protease appeared responsible for size variation from the predicted size. The activity and stability over broad range of temperature and pH, and substrate specificity also suggested its potential for use in therapeutic applications.
References
Bennett, G. N., & Hickford, J. G. H. (2011). Ovine footrot: New approaches to an old disease. Veterinary Microbiology, 148, 1–7.
Kennan, R. M., Gilhuus, M., Frosth, S., Seemann, T., Dhungyel, O. P., Whittington, R. J., Boyce, J. D., Powell, D. R., Aspan, A., Jorgensen, H. J., Bulach, D. M., & Rooda, J. I. (2014). Genomic evidence for a globally distributed. Bimodal Population in the Ovine Footrot Pathogen Dichelobacter nodosus. mBio, 5(5), e01821–14.
Egerton, J. R., Roberts, D. S., & Parsonson, I. M. (1969). The aetiology and pathogenesis of ovine footrotA histological study of the bacterial invasion. Journal of Comparative Pathology, 79, 209–216.
Stewart, D. J., Clark, B. L., & Jarret, R. G. (1984). Difference between strains of Bacteroides nodosus in their effects on the severity of footrot, bodyweight and wool growth on Merino sheep. Australian Veterinary Journal, 61, 349–352.
Ghosh, A., Chakrabarti, K., & Chattopadhyay, D. (2009). Cloning of feather-degrading minor extracellular protease from Bacillus cereus DCUW: dissection of the structural domains. Microbiology, 155, 2049–57.
Arnorsdottir, J., Smaradottir, R. B., Magnusson, O. T., Thorbjarnardottir, S. H., Eggertsson, G., & Kristjansson, M. (2002). Characterization of a cloned subtilisin-like serine proteinase from a psychrotrophic Vibrio species. European Journal of Biochemistry, 269, 5536–5546.
Kwon, K., Hasseman, J., Latham, S., Grose, C., Do, Y., Fleischmann, R. D., Pieper, R., & Petersen, S. N. (2011). Recombinant expression and functional analysis of proteases from Streptococcus pneumoniae, Bacillus anthracis and Yersinia pestis. BMC Biochemistry, 12, 17.
Larsen, A. N., Moe, E., Helland, R., Gjellesvik, D. R., & Willassen, N. P. (2006). Characterization of a recombinantly expressed proteinase K-like enzyme from a psychrotrophic Serratia sp. FEBS Journal, 273, 47–60.
Maurer, K. H. (2004). Detergent proteases. Current Opinion in Biotechnology, 15, 330–334.
Craik, C. S., Page, M. J., & Madison, E. L. (2011). Proteases as therapeutics. Biochemical Journal, 435, 1–16.
Chanalia, P., Gandhi, D., Jodha, D., & Singh, J. (2011). Applications of microbial proteases in pharmaceutical industry: an overview. Reviews in Medical Microbiology, 22, 96–101.
Shen, H. B., & Chou, K. C. (2009). Identification of proteases and their types. Analytical Biochemistry, 385, 153–160.
Skerman, T. M. (1975). Determination of some in vitro growth requirements of Bacteroides nodosus. Journal of General Microbiology, 87, 107–119.
La Fontaine, S., Egerton, J. R., & Rood, J. I. (1993). Detection of Dichelobacter nodosus using species specific oligonucleotides as PCR primers. Veterinary Microbiology, 35, 101–117.
Dhungyel, O. P., Whittington, R. J., & Egerton, J. R. (2002). Serogroup specific single and multiplex PCR with pre-enrichment culture and immuno-magnetic bead capture for identifying strains of D. nodosus in sheep with footrot prior to vaccination. Molecular and Cellular Probes, 16, 285–296.
Cheetham, B. F., Tanjung, L. R., Sutherland, M., Druitt, J., Green, G., McFarlane, J., Bailey, G. D., Seaman, J. T., & Katz, M. E. (2006). Improved diagnosis of virulent footrot using intA gene. Veterinary Microbiology, 116, 166–174.
Palmer, M. A. (1993). A gelatin test to detect activity and stability of proteases produced by Dichelobacter (Bacteroides) nodosus. Veterinary Microbiology, 36, 113–122.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., & Lipman, D. J. (1990). Basic local alignment search tool. Journal of Molecular Biology, 215, 403–410.
Letunic, C. R. R., Pils, B., Pinkert, S., Schultz, J., & Bork, P. (2006). SMART 7: recent updates to the protein domain annotation resource. Nucleic Acids Research, 34, D257–260.
Marchler-Bauer, A., Anderson, J. B., Cherukuri, P. F., DeWeese-Scott, C., Geer, L. Y., He, M. G. S., Hurwitz, D. I., Jackson, J. D., Ke, Z., Lanczycki, C. J., Liebert, C. A., Liu, C., Lu, F., Marchler, G. H., Mullokandov, M., Shoemaker, B. A., Simonyan, V., Song, J. S., Thiessen, P. A., Yamashita, R. A., Yin, J. J., Zhang, D., & Bryant, S. H. (2005). CDD: a Conserved Domain Database for protein classification. Nucleic Acids Research, 33, 192–196.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., & Kumar, S. (2011). MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Molecular Biology and Evolution, 28, 2731–2739.
Schwede, T., Kopp, J., Guex, N., & Peitsch, M. C. (2003). SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Research, 31, 3381–3385.
Benkert, P., Tosatto, S. C. E., & Schomburg, D. (2008). QMEAN: A comprehensive scoring function for model quality assessment. Proteins: Structure, Function, and Bioinformatics, 71(1), 261–277.
Folin, O., & Ciocalteu, V. (1927). On tyrosine and tryptophane determinations in proteins. Journal of Biological Chemistry, 73, 627–650.
Wang, Q., Miao, J. L., Hou, Y. H., Ding, Y., Wang, G. D., & Li, G. Y. (2005). Purification and characterization of an extracellular cold–active serine protease from the psychrophilic bacterium Colwellia sp. NJ341. Biotechnology Letters, 27, 1195–1198.
Rao, C. S., Sathish, T., Brahamaiah, P., Kumar, T. P., & Prakasham, R. S. (2009). Development of a mathematical model for Bacillus circulans growth and alkaline protease production kinetics. Journal of Chemical Technology and Biotechnology, 84, 302–307.
Kennan, R. M., Wong, W., Dhungyel, O. P., Han, X., Wong, D., Parker, D., Rosado, C. J., Law, R. H., McGowan, S., Reeve, S. B., Levina, V., Powers, G. A., Pike, R. N., Bottomley, S. P., Smith, A. I., Marsh, I., Whittington, R. J., Whisstock, J. C., Porter, C. J., & Rood, J. I. (2010). The Subtilisin-like protease AprV2 is required for virulence and uses a novel disulphide-tethered exosite to bind substrates. PLoS Pathogens, 6(11), E1001210.
Myers, G. S., Parker, D., Al-Hasani, K., Kennan, R. M., Seemann, T., Ren, Q., Badger, J. H., Selengut, J. D., Deboy, R. T., Tettelin, H., Boyce, J. D., McCarl, V. P., Han, X., Nelson, W. C., Madupu, R., Mohamoud, Y., Holley, T., Fedorova, N., Khouri, H., Bottomley, S. P., Whittington, R. J., Adler, B., Songer, J. G., Rood, J. I., & Paulsen, I. T. (2007). Genome sequence and identification of candidate vaccine antigens from the animal pathogen Dichelobacter nodosus. Nature Biotechnology, 25(5), 569–575.
Riffkin, M. C., Wang, L. F., Kortt, A. A., & Stewart, D. J. (1995). A single amino-acid change between the antigenically different extracellular serine proteases V2 and B2 from Dichelobacter. Gene, 167(1–2), 279–283.
Deng, A., Wu, J., Zhang, G., & Wen, T. (2011). Molecular and structural characterization of a surfactant-stable high-alkaline protease AprB with a novel structural feature unique to subtilisin family. Biochimie, 93, 783–791.
VanderLaan, J. M., Teplyakov, A. V., Kelders, H., Kalk, K. H., Misset, O., Mulleners, L. J., & Dijkstra, B. W. (1992). Crystal structure of the high-alkaline serine protease PB92 from Bacillus alcalophilus. Protein Engineering, 5(5), 405–411.
Wandersman, C. (1989). Secretion, processing and activation of bacterial extracellular proteases. Molecular Microbiology, 3(12), 1825–1831.
Wong, W., Kennan, R. M., Rosado, C. J., Rood, J. I., Whisstocka, J. C., & Portera, C. J. (2010). Crystallization of the virulent and benign subtilisin-like proteases from the ovine footrot pathogen Dichelobacter nodosus. Acta Crystallographica Section, F66, 289–293.
Rawlings, N. D., & Barett, A. J. (1994). Families of serine peptidases. Methods in Enzymology, 224, 19–61.
Ather, A. (2009). Identification, cloning and expressions of proteases from a cold adapted organism Aliivibrio salmonicida KJE-3900. University of Tromso.
Vaughan, P. R., Wang, L. F., Stewart, D. J., Lilley, G. G., & Kortt, A. A. (1994). Expression in Escherichia coli of the extracellular basic protease frorn Dichelobacter nodosus. Microbiology, 140, 2093–2100.
Sarvas, M., Harwood, C. R., Bron, S., & Dijl, J. M. (2004). Post-translocational folding of secretory proteins in Gram-positive bacteria. Biochimica et Biophysica Acta, 1694, 311–327.
Godde, C., Sahm, K., Brouns, S. J. J., Kluskens, L. D., Oost, J. V., deVos, W. M., & Antranikian, G. (2005). Cloning and expression of islandisin, a new thermostable subtilisin from Fervidobacterium islandicum, in Escherichia coli. Applied and Environmental Microbiology, 71(7), 3951–3958.
Chen, X. L., Xie, B. B., Lu, J. T., He, H. L., & Zhang, Y. Z. (2007). A novel type of subtilase from the psychrotolerant bacterium Pseudoalteromonas sp. SM9913: catalytic and structural properties of deseasin MCP-01. Microbiology, 153, 2116–2125.
Huston, A. L., Methe, B., & Deming, J. W. (2004). Purification, characterization, sequencing of an extracellular cold-active aminopeptidase produced by marine psychrophile Colwellia psychrerythraea strain 34H. Applied and Environmental Microbiology, 70, 2321–2328.
Kortt, A. A., Caldwell, J. B., Lilley, G. G., Edwards, R., Vaughan, J., & Stewart, D. J. (1994). Characterization of a basic serine protease (pI ∼ 9.5) secreted by virulent strains of Dichelobacter nodosus and identification of a distinct, but closely related, proteinase secreted by benign strains. Journal of Biochemistry, 299, 521–525.
Setyorini, E., Kim, Y. J., Takenaka, S., Murakami, S., & Aoki, K. (2006). Purification and characterization of a halotolerant intracellular protease from Bacillus subtilis strain FP-133. Journal of Basic Microbiology, 46(4), 284–304.
Yan, B. Q., Chen, X. L., Hou, X. Y., He, H., Zhou, B. C., & Zhang, Y. Z. (2009). Molecular analysis of the gene encoding a cold-adapted halophilic subtilase from deep-sea psychrotolerant bacterium Pseudoalteromonas sp. SM9913: cloning, expression, characterization and function analysis of the C-terminal PPC domains. Extremophiles, 13, 725–733.
Pushpam, P. L., Rajesh, T., & Gunasekaran, P. (2011). Identification and characterization of alkaline serine protease from goat skin surface metagenome. AMB Express, 1, 3.
Zhang, Y., Zhao, J., & Zeng, R. (2011). Expression and characterization of a novel mesophilic protease from metagenomic library derived from Antarctic coastal sediment. Extremophiles, 15, 23–29.
Doddapaneni, K. K., Tatineni, R., Potumarthi, R., & Mangamoori, L. N. (2007). Optimization of media constituents through response surface methodology for improved production of alkaline proteases by Serratia rubidaea. Journal of Chemical Technology and Biotechnology, 82, 721–729.
Morita, Y., Hasan, Q., Sakaguchi, T., Murakami, Y., Yokoyama, K., & Tamiya, E. (1998). Properties of a cold-active protease from psychrotrophic Flavobacterium balustinum P104. Applied Microbiology and Biotechnology, 50(6), 669–675.
Secades, P., Alvarez, B., & Guijarro, J. A. (2001). Purification and characterization of a psychrophilic calcium induced, growth-phase dependent metalloprotease from the fish pathogen Flavobacterium psychrophilum. Applied and Environmental Microbiology, 67(6), 2436–2444.
Tariq, A. L., Reyaz, A. L., & Prabakaran, J. J. (2011). Purification and Characterization of 56 KDa cold active protease from Serratia marcescens. African Journal of Microbiology Research, 5(32), 5841–5847.
Shankar, S., Rao, M., & Laxman, R. S. (2011). Purification and characterization of an alkaline protease by a new strain of Beauveria sp. Process Biochemistry, 46, 579–585.
Srilakshmi, J., Madhavi, J., Lavanya, S., & Ammani, K. (2015). Commercial potential of fungal protease: past, present and future prospects. Journal of Pharmaceutical, Chemical and Biological Sciences, 2(4), 218–234.
Acknowledgments
The authors acknowledge the financial support received from the ICAR under NAIP project on footrot. The authors also acknowledge CSK-Himachal Pradesh Agricultural University, Palampur, for logistic support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Rights and permissions
About this article

Cite this article
Wani, A.H., Sharma, M., Salwan, R. et al. Cloning, Expression, and Functional Characterization of Serine Protease Aprv2 from Virulent Isolate Dichelobacter nodosus of Indian Origin. Appl Biochem Biotechnol 180, 576–587 (2016). https://doi.org/10.1007/s12010-016-2117-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12010-016-2117-5