
Abstract
Using a previously created Mycobacterium avium genomic library with GFP-promoter trap in Mycobacterium smegmatis, we screened for genes that are upregulated upon infection of A. castellanii. Clones exhibiting a 2.5-fold or greater increase in GFP expression, out of a total of 10,000 clones, were selected for further examination. Upregulation was confirmed in subsequent experiments. A total of 20 clones showed an increase in expression 24 and 48 h after infection. Homologues were identified, and genes were found to encode for a variety of functions, including metabolic pathways, protein transcription and translation, and macromolecule degradation. Eight out of the 20 genes were found to be the same as those upregulated upon human macrophage infection. Five genes were selected to confirm upregulation in M. avium following amoeba infection, using real time PCR. All 5 genes were found to be upregulated at least 2.5-fold in M. avium. These results showed that the GFP promoter library in M. smegmatis is a valid system for studying gene upregulation in M. avium systems, and that many M. avium genes are commonly upregulated following macrophage and amoeba infection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Mycobacterium avium is an emerging opportunistic pathogen. It is known to cause infections in both humans and animals, being especially prevalent in immunocompromised individuals, such as HIV-infected patients [12]. M. avium infections are also found in patients with predisposing lung conditions, like emphysema, bronchiectasis, and cystic fibrosis [13, 20]. Other entities that are associated with increased risk include deficiency in interleukin-12 receptor [1], functional deficiency of interferon-gamma receptors, and immunosuppression due to transplantation [8, 18]. In both AIDS patients and those with other immunodeficiencies, infections are often disseminated.
M. avium is a ubiquitous organism, often found in soil and water sources. Studies of M. avium in water systems report a range of 10 to 700,000 CFU/l, with higher numbers in the distribution system than downstream [10]. Chlorine, the main agent of bacterial control in water systems, has been shown to have varying effects on M. avium, with some strains being less susceptible than others [9]. This characteristic may allow slow-growing M. avium strains to remain in drinking water systems from weeks to months.
Free-living amoeba are common inhabitants of water, including treated drinking water. One proposed theory is that, like Legionella pneumophila, M. avium may use amoeba as an intermediate host. It has been shown that M. avium will grow in co-culture with amoeba, often found within the outer walls of double-walled cysts of amoeba [19]. It has also been found in non-cystic amoeba in the environment (unpublished observation). Prior interaction between M. avium and amoeba was shown to enhance both entry and intracellular replication of M. avium in macrophages, and to be associated with a more virulent phenotype of M. avium in the beige mouse model [4].
Studies involving Legionella have indicated that genes that play a role in macrophage invasion and replication are often the same genes as those found with roles in amoeba invasion and replication. For instance, rtxA is involved in adherence and entry of macrophages and amoeba [5], and icmT, which is necessary for pore-formation mediated cytolysis [15], has a function associated with the ability of the bacteria to exit the macrophage or amoeba. The gene htrA, however, is necessary for intracellular replication in macrophages, but is not required for survival in amoeba [17].
Although studies have been carried out to determine M. avium genes upregulated upon infection of macrophages, no information exists regarding genes required during life within amoeba. Therefore, we undertook experiments with the goal of identifying M. avium genes upregulated during infection of amoeba.
Materials and Methods
Bacteria, amoeba, and growth conditions
M. smegmatis mc2 155 was kindly provided by Dr. William Jacobs, Jr. (Albert Einstein School of Medicine, NY). M. smegmatis was cultured on 7H10 agar, supplemented with oleic acid, albumin, dextrose, and catalase (OADC, Difco Laboratories, Detroit, MI) at 37°C. The strain was used for survival studies, as well as for the construction of the M. avium GFP-promoter library [6]. M. avium 104 strain was isolated from blood of an AIDS patient. It was cultured in 7H9 broth supplemented with OADC at 37°C. Acanthamoeba castellanii was obtained from ATCC (ATCC number 30234). It was maintained in 712 PYG media, in the dark, at room temperature.
Mycobacterium smegmatis and Mycobacterium avium: Ability to survive within A. castellanii
Due to the fact that the M. avium promoter GFP library was constructed in M. smegmatis, survivability of M. smegmatis in A. castellanii was determined. We also defined the ability of M. avium to survive in amoeba and the time points for RNA extraction of intracellular M. avium in A. castellanii. A. castellanii was seeded in 12-well flat bottom tissue culture plates at 1.0 × 108 cells, in 712 PYG media, at room temperature, and allowed to adhere overnight. Preparation of a disperse inoculum was carried out as previously described [4]. Established monolayers were infected with disperse 107 bacteria (M. smegmatis mc2 155 or M. avium) and incubated at either 30°C or 37°C for 2 h. A. castellanii monolayer was then washed 3 times with Hanks Balanced Salt Solution (HBSS) to remove extracellular bacteria, a method previously demonstrated to efficiently remove uningested bacteria [4]. At 0, 24, 48, 72, and 96 h time points, A. castellanii monolayer then was lysed with 0.25% SDS for 30 min (a concentration that has no effect on mycobacteria viability) and passed through a 26-gauge needle to ensure complete lysis of the amoeba. The lysate was plated onto 7H11 agar plates and grown for 10 days at 37°C to determine the number of colonies of intracellular M. smegmatis or intracellular M. avium.
M. avium promoter GFP library
Library construction has been reported previously [6]. Briefly, M. avium genomic DNA was extracted and digested with Sau3A. Fragments ranging from 300–1000 bp were selected using gel electrophoresis and cloned into plasmid pEMC1 [6]. Plasmids were expanded in E. coli, purified, and then transformed into M. smegmatis. Positive clones were selected using kanamycin 50 μg/ml on 7H10 agar. The pools containing 5 clones each were stored per well of a 96-well plate. A total of 10,000 clones, representing approximately 2.0-times the size of the M. avium genome, were used.
Infection of A. castellanii with library
Amoeba in 712 PYG medium were seeded in a 96-well flat-bottom tissue culture plate at 1.0 × 105 cells per well, and allowed to adhere overnight at room temperature in the dark. Resultant monolayers were infected with 106 bacteria (MOI 10) and allowed to incubate at 30°C for 2 h. To remove extracellular bacteria, amoeba were washed 3 times with HBSS and media was replenished [4]. GFP expression was measured prior to infection to establish a baseline and at 4, 24, and 48 h after infection, using a Cytofluorimeter II (Biosearch, Bedford, MA). Wells exhibiting at least a 2-fold increase in the level of GFP over baseline were lysed with 0.25% SDS and plated onto 7H11 agar plates with 50 μg/ml kanamycin. Twenty individual clones per well were selected from plates and cultured in 7H9 Middlebrook broth containing 50 μg/ml kanamycin. They were used to infect fresh amoeba following the same procedure described above, with expression of GFP being measured at the same time points of 4, 24, and 48 h. Individual clones exhibiting at least a 2.5-fold increase in GFP expression over baseline were selected for sequencing. The selected clones were propagated in 7H9 broth with 50 μg/ml kanamycin, and plasmids were extracted using the Stratagene Plasmid Extraction Kit, as recommended by the manufacturer (Stratagene, San Diego, CA). Plasmids were then transformed, using electroporation, into E. coli. The E. coli was grown in LB broth in the presence of 50 μg/ml kanamycin. Plasmids were again extracted and sent for sequencing with primers specific for the GFP and kanamycin sequences in the pEMC1 plasmid.
Sequencing and analysis
Sequencing was performed by the Center for Gene Research and Biotechnology, Oregon State University. Sequence analysis was performed using the BLAST service provided by the National Center for Biotechnology Information (www.ncbi.nih.gov). To determine the M. avium gene function, we searched for either M. tuberculosis or M. avium ssp. paratuberculosis homologues to the genes upregulated in our study. This was done using Tuberculist (http://genolist.pasteur.fr/TubercuList/) and BLAST.
RNA extraction and isolation
M. avium grown in 7H9 broth was used as a control, and intracellular M. avium from infected amoeba was obtained for experimental RNA. Amoeba were infected as described above, using M. avium strain 104. At 24 h, amoeba were lysed with 0.25% SDS for 30 min and passed through a 23-gauge needle to insure complete breakage of amoeba. Resulting lysate was centrifuged at 400× rpm for 5 min to removed lysed amoeba from suspension. Supernatant was centrifuged at 3200× rpm, and the resulting pellet was used for obtaining RNA. RNA was extracted using a series of phenol/chloroform extractions as previously described [6]. RNA was cleaned using an RNA clean kit and treated with DNAse I prior to real-time PCR. Integrity of RNA was confirmed by measuring OD260/280nm absorption and gel electrophoresis. Total RNA was reverse transcribed with Superscript II Plus RNase H- Reverse Transcriptase (Invitrogen, Carlsbad, CA) per the manufacturer’s instructions.
Real time PCR
SYBR green technology was used to perform real time PCR. Run protocol for the LightCycler was as follows: cycle 1, denaturation step, 95°C for 10 min; cycle 2, amplification and quantation, repeated 40 times, 95°C for 30 s, 63°C for 30s, and 72°C for 2 min with one fluorescence measurement. Threshold cycle (Ct) was quantified as described in User Bulletin #2 for ABI PRIMS 7700 sequence detection system (ABI). Threshold cycle is defined as the fractional cycle number at which the fluorescence reaches 10× the standard deviation of the baseline. iCycler I software (BIORAD, Hercules, CA) was used for quantitative analysis, and a relative quantification was used in which the expression levels of M. avium target genes were compared to a standard curve generated using several dilutions of a known quantity of amplicons. As previously described [6], an amplification-based strategy was used to determine fold change in gene expression. For each gene amplification, before calculating fold change the Ct values were normalized to the Ct of the 16sRNA. The following formula was used: fold change = 2−Δ(ΔCt) where (ΔCt) = Ct, target – Ct, 16sRNA, Δ(ΔCt) = ΔCt, expressed – ΔCt, control.
Standard deviations were calculated, and samples were compared to values for gene MA2610c, which was used as a negative control because it is not up-regulated upon M. avium infection of amoeba.
Results
M. smegmatis and M. avium: Ability to survive within A. castellanii
To analyze the ability of M. smegmatis and M. avium to survive in amoeba, we determined CFU/ml at 4, 24, 48, 72, and 96 h after inoculation, at temperatures of 30°C and 37°C. As demonstrated in Figure 1, both M. smegmatis and M. avium showed the ability to survive within amoeba at both temperatures, although it is evident that M. smegmatis did not seem to replicate after 2 days at both 30°C and 37°C. However, at 37°C, a significant number of amoeba were noted to become detached from the bottoms of the wells. In addition, at 37°C an amoeba is not very effective in killing intracellular bacteria. For these reasons, we chose 30°C for the temperature of incubation of A. castellanii infected with the M. smegmatis GFP-promoter library and for infection of A. castellanii with M. avium.

M. smegmatis and M. avium growth in A. castellanii, both at 30°C and 37°C. Data points represent the average of 4 experiments ± SD.
M. avium gene expression determined by GFP library
Over 10,000 clones of the M. avium promoter library were screened for increased GFP expression. Twenty clones were identified as having a 2.5-fold or greater increase in expression of GFP (Table 1). Five genes showed upregulation only at the 24-h time point. Nine genes showed upregulation only at the 48-h time point. Six genes showed upregulation at both the 24- and 48-h time points. There was no significant increase in GFP expression at the 4-h time point over baseline. The genes identified belong to a variety of classes including transcription regulation, protein translation, metabolic pathways, energy metabolism, degradation of macromolecules, DNA replication, membrane proteins, and genes with unknown function. One gene of unknown function (clone 245) was found to be in a region that shared very little homology with other closely related bacteria such as M. tuberculosis or M. avium ssp. paratuberculosis. It also had no homology with other bacteria. This region was found to occupy 50 kb of the M. avium genome, and to have a G+C content of 69%, which is similar to the G+C content of M. avium.
Analysis of M. avium gene expression in amoeba by quantitative real time PCR
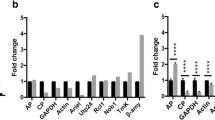
Five genes were chosen for analysis by real time PCR (Table 2). This allowed us to confirm upregulation of genes identified in the M. smegmatis system in M. avium, and would validate the use of the M. smegmatis library. The following M. avium genes were chosen for analysis: Ma0359 (membrane protein), Ma-lipL (esterase), Ma-embB (cell wall biosynthesis), Ma-lprC (lipoprotein), and Ma-nirB (nitrate reductase flavoprotein). These represent a variety of classes found to be upregulated in the GFP-promoter library. The gene Ma2610c did not show upregulation in amoeba using the promoter library. This gene served as a negative control.
Real-time PCR efficiency was determined using a dilution series of cDNA template with a fixed concentration of primers. Slopes calculated by the LightCycler software were used with the following formula to calculate efficiency: E = 10(−1/slope). These calculations indicated both a high real time efficiency with a high linearity.
Because expression of 16sRNA is constant independent of conditions, target genes from both control and experimental groups were normalized to the expression level of the 16sRNA gene. All 5 genes showed significant induction upon M. avium infection of amoeba, while no upregulation was observed with Ma2610c, the negative control, confirming that genes detected using the GFP promoter library were expressed by M. avium (Fig. 2).

Upregulation of M. avium genes upon infection of A. castellanii, as determined by real-time PCR. The data represents the average of 2 independent experiments ± SD.
Discussion
M. avium is an organism commonly encountered in the environment, mainly in soil and water sources [10, 12], where it shares the environment with other bacteria, as well as protozoa. Recently, M. avium has been shown to infect and survive in acantamoeba [4], and there is evidence suggestive that it is associated with the upregulation of genes associated with virulent phenotype of the bacterium. To seek a better understanding of the bacterial response to the intracellular environment in Acanthamoeba, we examine how amoeba infection impacts the regulation of M. avium genes.
Genes found to be upregulated in this study span a wide variety of classes. These include metabolic pathways, transcriptional regulators, DNA replication, energy metabolism, protein translation and modification, and degradation of macromolecules. Several of the genes seen to be upregulated upon Acanthamoeba infection have an unknown function. A number of the genes only show upregulation after 24 h of infection but not at the 4-h time point. This can be explained by an initial period of adaptation following uptake, in which many bacteria stop multiplying. It is very likely that this period of adaptation lasts less than 24 h. Among the genes upregulated is a promoter found in a region that has no homology to bacterial sequences stored in the database (clone 2H5). Close examination of the region found a 50-kb section of the M. avium genome that does not have significant homology to M. tuberculosis or M. avium ssp. paratuberculosis. The region has a G+C content of 69%, which suggests that the sequence either belongs to mycobacteria or has been acquired from another organism with high G + C content in the genome. Alternatively, the region may have been lost in both M. tuberculosis and M. paratuberculosis. This region, therefore, might be an evolutionary adaptation that is required for M. avium to be able to infect amoeba but probably not macrophages. In fact, this region has not been shown to be upregulated in macrophages, despite some similarities between amoeba and macrophage vacuoles [3, 4, 7, 14].
Eight of the upregulated genes found in this study were the same as those identified in infection of macrophages by M. avium [6, 11]. These included genes involved in protein translation, energy metabolism, and genes of unknown function, suggesting that M. avium uses some mechanisms for survival in the intracellular environment that are common for amoeba and macrophage. Studies with Legionella pneumophila suggested that Legionella expresses many of the same virulence genes when within macrophages and amoeba [5, 15]. In contrast, some virulence genes are expressed exclusively in one environment. For example, the htrA is a necessary gene for full virulence in macrophages but is not needed in amoeba [17]. This appears to be the case of the M. avium gene in the 50-kb exclusive region, which has not been shown to be expressed within macrophages in several studies.
The nirB gene has been shown to be over-expressed in intracellular M. avium by both Hou and Danelishvili. This gene is part of the denitrification process, encoding for a nitrogen reductase. Genes in this category, such as nirK and nirV, have been shown to regulate in virulence in Brucella in mice [2]. Nir is required for anaerobic growth and plays a role against NO toxicity in macrophages.
The lprC is another gene in common between macrophage and amoeba infection. This gene encodes for a lipoprotein, which may have a role in bacteria binding to host proteins, and in envelope remodeling [6]. Also upregulated were a ferredoxin reductase (Ma0688) and several dehydrogenases (serA2 and MAPfadE25 homologue). Ferredoxin reductase is an enzyme that catalyzes the oxidation and reduction of ferredoxin or adrenodoxin in the presence of NADP. Iron starvation response in Pseudomonas aeruginosa has been shown to lead to a cascade that not only activates ferredoxin, ferredoxin reductase, and several dehydrogenases, but also activates genes encoding virulence factors such as TonB and exotoxinA [16]. M. avium phagosome has been shown to be depleted in iron initially, but to recruit iron after a few hours [21].
Using an M. avium GFP promoter library, we have identified 20 genes upregulated during infection of amoeba. Up-regulation of 5 of those genes was then confirmed by real-time PCR, confirming that the system can be used in a selective manner. Many genes expressed have unknown function, but future studies involving inactivation of these genes may provide information on their roles in the pathogenesis of the infection. Although not complete, similarities are likely to exist between the M. avium environment in amoeba and macrophages.
Literature Cited
Altare F, Durandy A, Lammas D, Emile JF, Lamhamedi S, Le Deist F, Drysdale P, Jouanguy E, Doffinger R, Bernaudin F, Jeppsson O, Gollob JA, Meinl E, Segal AW, Fischer A, Kumararatne D, Casanova JL (1998) Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science 280:1432–1435
Baek SH, Rajashekara G, Splitter GA, Shapleigh JP (2004) Denitrification genes regulate Brucella virulence in mice. J Bacteriol 186:6025–6031
Brown RC, Bass H, Coombs JP (1975) Carbohydrate binding proteins involved in phagocytosis by Acanthamoeba. Nature 254:434–435
Cirillo JD, Falkow S, Tompkins LS, Bermudez LE (1997) Interaction of Mycobacterium avium with environmental amoebae enhances virulence. Infect Immun 65:3759–3767
Cirillo SL, Yan L, Littman M, Samrakandi MM, Cirillo JD (2002) Role of the Legionella pneumophila rtxA gene in amoebae. Microbiology 148:1667–1677
Danelishvili L, Poort MJ, Bermudez LE (2004) Identification of Mycobacterium avium genes up-regulated in cultured macrophages and in mice. FEMS Microbiol Lett 239:41–49
Davies B, Edwards SW (1991) Chemiluminescence and superoxide production in Acanthamoeba castellanii: free radicals generated during oxidative stress. J Gen Micro 137:1021–1027
Dorman SE, Picard C, Lammas D, Heyne K, van Dissel JT, Baretto R, Rosenzweig SD, Newport M, Levin M, Roesler J, Kumararatne D, Casanova JL, Holland SM (2004) Clinical features of dominant and recessive interferon gamma receptor 1 deficiencies. Lancet 364:2113–2121
Falkinham JO 3rd (2003) Factors influencing the chlorine susceptibility of Mycobacterium avium, Mycobacterium intracellulare, and Mycobacterium scrofulaceum. Appl Environ Microbiol 69:5685–5689
Falkinham JO 3rd, Norton CD, LeChevallier MW (2001) Factors influencing numbers of Mycobacterium avium, Mycobacterium intracellulare, and other Mycobacteria in drinking water distribution systems. Appl Environ Microbiol 67:1225–1231
Hou JY, Graham JE, Clark-Curtiss JE (2002) Mycobacterium avium genes expressed during growth in human macrophages detected by selective capture of transcribed sequences (SCOTS). Infect Immun 70:3714–3726
Inderlied CB, Kemper CA, Bermudez LE (1993) The Mycobacterium avium complex. Clin Microbiol Rev 6:266–310
Kilby JM, Gilligan PH, Yankaskas JR, Highsmith WE Jr, Edwards LJ, Knowles MR (1992) Nontuberculous mycobacteria in adult patients with cystic fibrosis. Chest 102:70–75
Lock R, Oehman L, Dahlgren C (1987) Phagocytic recognition mechanisms in human granulocytes and Acanthamoeba castellanii using type 1 fimbriated Escherichia coli as phagocytic prey. FEMS Microbiol Lett 44:135–140
Molmeret M, Alli OA, Zink S, Flieger A, Cianciotto NP, Kwaik YA (2002) icmT is essential for pore formation-mediated egress of Legionella pneumophila from mammalian and protozoan cells. Infect Immun 70:69–78
Ochsner UA, Wilderman PJ, Vasil AI, Vasil ML (2002) GeneChip expression analysis of the iron starvation response in Pseudomonas aeruginosa: identification of novel pyoverdine biosynthesis genes. Mol Microbiol 45:1277–1287
Pedersen LL, Radulic M, Doric M, Abu Kwaik Y (2001) HtrA homologue of Legionella pneumophila: an indispensable element for intracellular infection of mammalian but not protozoan cells. Infect Immun 69:2569–2579
Simpson GL, Raffin TA, Remington JS (1982) Association of prior nocardiosis and subsequent occurrence of nontuberculous mycobacteriosis in a defined, immunosuppressed population. J Infect Dis 146:211–219
Steinert M, Birkness K, White E, Fields B, Quinn F (1998) Mycobacterium avium bacilli grow saprozoically in coculture with Acanthamoeba polyphaga and survive within cyst walls. Appl Environ Microbiol 64:2256–2261
Teirstein AS, Damsker B, Kirschner PA, Krellenstein DJ, Robinson B, Chuang MT (1990) Pulmonary infection with Mycobacterium avium-intracellulare: diagnosis, clinical patterns, treatment. Mt Sinai J Med 57:209–215
Wagner D, Maser J, Lai B, Cai Z, Barry CE 3rd, Honer Zu Bentrup K, Russell DG, Bermudez LE (2005) Elemental analysis of Mycobacterium avium-, Mycobacterium tuberculosis-, and Mycobacterium smegmatis-containing phagosomes indicates pathogen-induced microenvironments within the host cell’s endosomal system. J Immunol 174:1491–1500
Acknowledgments
We thank Denny Weber for preparing the manuscript. We are also indebted to the members of the Bermudez laboratory for their support and help with the experiments described. This work was supported in part by NIH grant AI-43199.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Tenant, R., Bermudez, L.E. Mycobacterium avium Genes Upregulated Upon Infection of Acanthamoeba castellanii Demonstrate a Common Response to the Intracellular Environment. Curr Microbiol 52, 128–133 (2006). https://doi.org/10.1007/s00284-005-0218-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00284-005-0218-4