
Abstract
Escherichia coli MazF is a toxin protein that cleaves RNA at ACA sequences. Its activation has been thought to cause growth inhibition, primarily through indiscriminate cleavage of RNA. To investigate responses following MazF activation, transcriptomic profiles of mazF-overexpressing and non-overexpressing E. coli K12 cells were compared. Analyses of differentially expressed genes demonstrated that the presence and the number of ACA trimers in RNA was unrelated to cellular RNA levels. Mapping differentially expressed genes onto the chromosome identified two chromosomal segments in which upregulated genes formed clusters, and these segments were absent in the chromosomes of E. coli strains other than K12. These results suggest that MazF regulates selective, rather than indiscriminate, categories of genes, and is involved in the regulation of horizontally acquired genes. We conclude that the primary role of MazF is not only cleaving RNA indiscriminately but also generating a specific cellular state.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bacterial chromosomes encode various toxin–antitoxin (TA) systems. For example, the chromosome of the Escherichia coli MG1655 strain, a derivative of the K12 linage, encodes at least 37 TA systems (Yamaguchi and Inouye 2011; Wang et al. 2012). The function of a toxin-encoding gene is repressed by the action of a cognate antitoxin gene in vegetatively growing cells but is de-repressed (activated) under certain conditions to cause complete cell death, reversible growth arrest, or both (Van Melderen and Saavedra De Bast 2009; Tripathi et al. 2014).
TA systems are classified into six types (type I–type VI) according to their mode of action (Aakre et al. 2013; Masuda et al. 2012; Yamaguchi and Inouye 2011; Wang et al. 2012). In type II TA systems, which have been studied the most, toxin proteins cause dysfunction in essential cellular processes, and cognate antitoxin proteins repress the actions of toxins through direct protein–protein interactions. Under conditions of stress, antitoxins of type II TA systems are degraded by stress-induced proteases, and consequently toxins are activated. mazEF is one of the most widely distributed type II TA systems in bacteria. The E. coli mazF gene encodes a toxin that is an ACA-specific endoribonuclease (RNA interferase). MazF activation has been thought to cause growth inhibition through indiscriminate cleavage of RNA, because 96% of E. coli coding sequences have at least one ACA sequence (Venturelli et al. 2017).
However, how growth inhibition is evoked following MazF activation remains unclear at the molecular level. To address this issue, omics analyses of mazF-overexpressing cells have been carried out. Typical RNA-seq and Poly-seq (polysome fractionation coupled with RNA-seq) analyses revealed that the activation of MazF leads to a reduction in the translation of the genes involved in cell metabolism and energy supply, as well as the selective synthesis of the "MazF-regulon" proteins (Sauert et al. 2016). In contrast, other transcriptome and proteome analyses indicated the non-selective synthesis of MazF-regulon proteins but the overall inhibition of protein synthesis after MazF activation (Culviner and Laub 2018; Mets et al. 2019). Time-series RNA-seq measurements of MazF-induced cells (2–8 min) showed the inhibition of cell division and specific upregulation of 11 TCA cycle enzymes (Venturelli et al. 2017). However, we assume that these studies lacked satisfactory biological replicates and/or bioinformatic analysis. In this study, we compared the transcriptomic profile of mazF-overexpressing cells to that of control non-expressing cells based on three biological replicates. From the results, many differentially expressed genes (DEGs) were detected, and bioinformatic analyses of DEGs highlighted new biological roles for MazEF.
Materials and methods
E. coli strains and general techniques
Wild-type E. coli K12 MG1655 (obtained from the National Bio Resource Research Project: NBRP, NIG, Japan) was used as the host strain. Cells were cultured in Luria broth in the presence or absence of appropriate antibiotics at 37 °C, unless otherwise stated. When expressing mazF from plasmids, transformants were pre-grown overnight, diluted 1:400 with fresh Luria broth, and cultured for 1.7 h. Next, l-arabinose was added to the medium at a final concentration of 0.1% to induce gene expression from plasmids.
Plasmid construction
To construct pBAD-mazF, a double-stranded DNA fragment containing the ribosomal-binding sequence (RBS) was prepared by hybridizing two complementary synthetic oligonucleotides (sGEM12, 5′-ctagagaaagaggagaaatactagtagcggccgctgca; sGEM13, 5′-gcggccgctactagtatttctcctctttct). The hybridized fragment was cloned into the SpeI–PstI moiety of pSB1C3-BAD (parts ID BBa_I0500, iGEM Foundation, Cambridge, MA), yielding pBAD-RBS/C. The pBAD-RBS/C plasmid was treated with EcoRI and PstI, and the resulting fragment containing the RBS was cloned into the EcoRI–PstI moiety of pSB6A1 (iGEM foundation), yielding pBAD-RBS. A DNA fragment containing the mazF coding sequence was PCR-amplified from MG1655 total DNA using a specific primer set (sGEM14, 5′-aaatctagatggtaagccgatacgtacccgatat and sGEM15, 5′-aaactgcagcggccgctactagtattattacccaatcagtacgttaattttggctttaatg). The fragment was treated with XbaI and PstI and cloned into the SpeI–PstI moiety of pBAD-RBS yielding pBAD-RBS-mazF/C. Then, the mazF-containing fragment was excised from pBAD-RBS-mazF/C using EcoRI and PstI and cloned into the EcoRI–PstI moiety of pSB6A1, yielding pBAD-mazF.
To construct plac-mazE, a DNA fragment containing lac promoter–RBS was excised from plac-RBS/C (parts ID BBa_J04500, iGEM Foundation) using EcoRI and PstI and cloned into the EcoRI–PstI moiety of pSB3K3 (iGEM Foundation), yielding plac-RBS. A DNA fragment containing the mazE coding sequence was PCR-amplified from MG1655 total DNA using a specific primer set (sGEM16, 5′-aaatctagatgatccacagtagcgtaaagcgttg and sGEM17, 5′-aaactgcagcggccgctactagtattattaccagacttccttatctttcggctctc). The fragment was treated with XbaI and PstI and cloned into the SpeI–PstI moiety of plac-RBS/C, yielding plac-RBS-mazE/C. Then, plac-RBS-mazE/C was treated with EcoRI and PstI and the resulting mazE-containing fragment was cloned into the EcoRI–PstI moiety of plac-RBS, yielding plac-mazE.
RNA extraction and sequencing by next generation sequencing (RNA-seq)
Before harvesting, cells were treated with RNAprotect Bacteria Reagent (Qiagen, Valencia, CA) to stabilize RNA. Total RNA was extracted using the acid hot phenol method (Aiba et al. 1981) and purified using an RNeasy Mini Kit (Qiagen) according to the manufacturer’s protocol. The MICROBExpress Bacterial mRNA Enrichment kit (Ambion, Austin, TX) was applied to total RNA to remove excess rRNA. cDNA libraries were constructed using a TruSeq Stranded mRNA and Total RNA Sample Preparation Kit (Illumina Inc., San Diego, CA) and sequenced with a MiSeq sequencer (Illumina) in paired-end-read mode for 2 × 75 cycles. Each sample was analyzed in triplicate. To obtain the read number, all samples were sequenced with Miseq twice.
Sequence analysis
High-quality RNA-seq reads were filtered using the trimmomatic program version 0.36 (Bolger et al. 2014) with the following parameters: ‘SE CROP:68 HEADCROP:7 MINLEN:18’. Reads were mapped to the chromosome by the EDGE-pro program version 1.3.1 (Magoc et al. 2013) using default settings. The numbers of mapped reads were 3,676,809, 6,611,324, and 5,350,605 for experiments on control cells and 5,002,966, 5,112,654, and 5,354,611 for experiments using mazF-overexpressing cells. Transcriptome profiles were compared using the edgeR package of R (McCarthy et al. 2012) with the exactTest mode. The genome index, gene annotation, and genome-sequence files were downloaded from the RefSeq web site (MG1655 GCA_000005845.2_ASM584v2 version). The Biostrings package of R (Ellrott et al. 2002) was used to count the number of ACA trimers in individual genes from a multi-fasta formatted file of the chromosomal sequence. To visualize the circular chromosome, the cgview program was used (Stothard and Wishart 2005).
RNA-seq data has been deposited in DDBJ/ENA/GenBank under Bioproject number PRJDB5742 and BioSample number SAMD00079815–SAMD00079832.
Results
Identification of DEGs and the number of ACA trimers following mazF activation
To express mazF in an inducible manner, a plasmid harboring the l-arabinose-inducible bad promoter and the mazF gene was constructed (pBAD-mazF). As expected, growth of the transformant with pBAD-mazF was inhibited upon the addition of l-arabinose (Supplementary Fig. S1). Besides, this phenotype was rescued by the 20-min delayed induction of mazE from the plasmid plac-mazE (Supplementary Fig. S1).
To investigate the cellular response following mazF activation, transcriptome profiles were compared between mazF-overexpressing and non-expressing cells. Cells harboring pBAD (an empty vector) or pBAD-mazF were cultured and treated with l-arabinose for 20 min, and RNA from these cells was subjected to RNA-seq analysis. When DEGs were defined as a log2-fold change > 2.5 and with false discovery rate (FDR) < 1.0 × 10–2, 266 and 245 genes were found to be up- and down-regulated, respectively, among 4318 genes (the full data set is shown in Supplementary Table S1). Relative levels of mazF mRNA increased by 54-fold with an FDR of 1.3 × 10–6.
For the first transcriptome analysis, the numbers of ACA trimers in each gene were enumerated for the total, upregulated, and down-regulated gene sets (Table 1). Assuming that MazF has an enhanced opportunity to cleave ACA-rich RNA, compared to that with ACA-less RNA and that the cleaved RNA is further rapidly degraded by RNases, ACA-less RNA, and short-length RNA would be enriched in the upregulated gene set. Likewise, opposite effect should be observed in down-regulated genes. However, no such enrichment was observed, and especially, RNA species with no ACA sequence were not enriched in the upregulated gene set. Therefore, RNA levels following MazF activation are not determined by the presence of ACA trimers. In contrast, a preceding study showed that the number of ACA trimers was negatively correlated with the mean log2-fold change in RNA abundance 8 min after the overexpression of MazF; however, such correlations were only observed for RNA species with fewer than three ACA trimers, and no correlations were observed after 2 min of overexpression (Venturelli et al. 2017). We then calculated the mean log2-fold change in RNA abundance and similarly investigated the correlation using our data (20-min overexpression), again showing no correlation (Supplementary Table S1). Altogether, the presence and number of ACA trimer determines RNA levels following MazF activation only under very specific conditions.
Biased distribution of DEGs on the chromosome
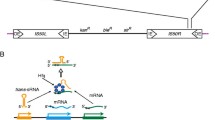
When mapping DEGs onto the E. coli circular chromosome, we found an uneven distribution (Fig. 1). In the chromosomal segments of 200–440 (named segment I) and 4480–4600 (segment II) kbp, a cluster of upregulated genes was formed, and no down-regulated genes were included. We further noticed that these two segments coincided with the specific segments reported by Karcagi et al. (2016); they found the segments that were present in the E. coli K12 MG1655 strain but absent in five other E. coli strains and deleted genes in these segments to construct a genome-streamlining strain MDS69 (the reconstructed genome map is shown in Supplementary Fig. S2). It should be noted that these segments contained only nonessential genes for growth and were assumed to be DNA containing genes for toxins, virulence factors, and mobile genetic elements that were recently acquired by horizontal transmission (Karcagi et al. 2016). Although the chromosomal segments of 1390–1480 kbp (segment III) also included a cluster of deleted genes in the MDS69 strain (Supplementary Fig. S2), this segment did not contain a cluster of upregulated genes (Fig. 1).

Chromosomal mapping of MazF-regulated genes in E. coli K12 MG1655. The black lines and characters indicate upregulated genes, and the brown lines and characters indicate downregulated genes. The full data set used for mapping is shown in Supplementary Table S2 (color figure online)
Meanwhile, when growth-essential genes (302 genes) (Yamazaki et al. 2008) were mapped similarly, no or few were found in segments I–III (Fig. 1; the reconstructed genome map is shown in Supplementary Fig. S3). Of 266 and 245 up- and down-regulated genes, two and 30 were growth-essential genes, respectively, and 88 and 26 genes were deleted genes in the MDS69 strain, respectively. Collectively, we conclude that MazF regulates gene expression non-uniformly. Clusters of strain-specific genes are upregulated, possibly reflecting involvement of MazF in the activation of horizontally acquired genes. In contrast, growth-essential genes tend to be down-regulated. To our knowledge, this is the first report to show that MazF-regulated genes form clusters in specific chromosomal segments.
Categorization of DEGs according to function
For an overview, DEGs and all genes were categorized into functional groups according to the KEGG Orthology (KO) database (Table 2) (Kanehisa et al. 2012). The most remarkable change was observed for the energy metabolism category; only one gene in this category (0.78%) was upregulated, whereas 42 genes (33%) were down-regulated. Moreover, in the carbohydrate metabolism category, 23% of genes were down-regulated. These results suggest that cells affirmatively limit energy acquisition from carbohydrates after MazF activation, and consequently, growth inhibition might occur. Similar results have been obtained by others, which have revealed that the activation of MazF leads to the downregulation of process associated with metabolism and energy supply (Sauert et al. 2016).
Enriched cellular pathways in DEGs
In the preceding section, DEGs were categorized into rough functional groups. To categorize DEGs and interpret the RNA-seq results in greater detail, gene ontology (GO) enrichment analysis was implemented using DAVID (Database for Annotation, Visualization and Integrated Discovery) with the default settings (Huang et al. 2009). From the resulting annotation list, the enriched cellular pathways (KEGG Pathways) were extracted, with a p value < 0.05 considered significant (Table 3). This analysis revealed that with respect to the upregulated genes (1) thiamine metabolism, (2) valine, leucine, and isoleucine biosynthesis, and (3) 2-oxocarboxylic acid metabolism pathways were enriched. The latter two pathways are closely related, and both are involved in branched chain amino acids biosynthesis. However, the biological role of this upregulation is unclear.
With respect to the downregulated genes, the result again suggests that cells are limited in acquiring energy from carbohydrate following MazF activation, and this limitation is accomplished at multiple metabolic pathways such as glycolysis, TCA cycle, oxidative phosphorylation, and sugar uptake (PTS) (Table 3). Besides, 11 genes in the nitrogen metabolism pathway, glnA, narKGHJI, napAB, nirBD, and nrfA, were downregulated (Table 3). These genes are all involved in nitrate reduction to ammonia and following glutamine synthesis, and thus, energy acquisition is also limited at the nitrate respiration step. However, Venturelli et al. reported that 11 TCA cycle genes were upregulated between 2 and 8 min after the induction of MazF expression (Venturelli et al. 2017). The difference in induction time (20 min in this study) might be the cause of these conflicting results.
Enriched gene categories in DEGs
The result of the above GO enrichment analysis was inspected further on enriched gene categories. Regarding the upregulated genes (Table 4), the GO annotation cluster related to transposon function showed the highest enrichment score. Furthermore, we manually found that, out of 25 genes of the CP4-6 cryptic prophage, 10 genes were upregulated, whereas no gene was down-regulated. The CP4-6 prophage is in the middle of the segment I (from 262 to 296 kb) (Ferenci et al. 2009; Wang et al. 2010), and these 10 genes are part of the upregulated gene cluster (Fig. 1). Similarly, out of 30 genes of the Qin cryptic prophage, six and one genes were up- and down-regulated, respectively. The Qin prophage belongs to none of the segments I–III. These results again suggest activation of horizontally acquired and mobile genetic elements, following MazF activation.
The annotation cluster related to the fimbria/pilus function was found to be upregulated (Table 4). Type 1 fimbriae of E. coli facilitate attachment to biotic and abiotic surfaces (Blumer et al. 2005), and multiple TA systems including MazEF play important roles in biofilm formation through the regulation of fimbria production (Kim et al. 2009; Kolodkin-Gal et al. 2009). Therefore, this upregulation contributes to biofilm formation, which helps bacteria survive harsh conditions.
Regarding down-regulated genes (Table 5), the functional categories mentioned in Table 3 were again listed. In addition, the annotation cluster related to chaperon function was listed, and groSL, dnaKJ, grpE, lsrR, hslUV, htpG, pykF, hslR, sodA, secB, hdeAB, ridA, and narJ were included in this annotation cluster. The expression of some chaperone genes including ibpAB, hslUV, and dnaKJ is known to be induced upon deletion of the mqsR toxin-encoding gene or the overexpression of mqsR for 15 min (Kim et al. 2010). These facts and our results both suggest that some toxin proteins regulate the expression of chaperone network genes both negatively and positively. As groSL, dnaKJ, grpE, hslUV, htpG, and hslR are members of the σ32 (sigma factor 32)-regulon (Nonaka et al. 2006), these genes might be downregulated through σ32. It should be noted that MazF might regulate the levels of chaperone-function genes directly and independently through its RNA interferase activity (see “Discussion” section for detail).
Crosstalk of MazF to other TA systems
It is well known that activation of one TA system causes sequential or simultaneous activation of other TA system(s) and that free toxins (toxins unbound to cognate antitoxins) can enhance transcription from other TA operons (Kasari et al. 2013). Considering these facts, transcriptional regulation of other TA systems was inspected in our RNA-seq data.
Out of 37 known TA systems, expression of six toxin and one antitoxin genes was upregulated, while expression of two toxin and one antitoxin genes was downregulated (Table 6). In these upregulated genes, two RNA interferase genes, higB and yafO, were included. Upregulation of higB (log2-fold change = 3.2, FDR = 6.1 × 10–3) did not accompany simultaneous upregulation of the cognate antitoxin gene, higA (log2-fold change = 1.4, FDR = 2.9 × 10–1), from which the ratio of toxin gene upregulation over antitoxin gene upregulation was calculated as 3.5. Upregulation of yafO (log2-fold change = 4.0, FDR = 6.9 × 10–4) accompanied elevated expression of cognate antitoxin gene yafN (log2-fold change = 2.5, FDR = 3.3 × 10–2; though this FDR value indicates insignificant upregulation), and similarly, the upregulation ratio was calculated as 2.8. Elevated expression of the other toxin gene, yafQ, was observed (log2-fold change = 2.3, FDR = 4.5 × 10–2; though this FDR value indicates insignificant upregulation) without simultaneous upregulation of cognate antitoxin gene dinJ (log2-fold change = 0.46, FDR = 7.6 × 10–1), and similarly, the upregulation ratio was calculated as 3.6. Taken together, growth inhibition by mazF overexpression may be caused through cleavage of RNAs with multiple RNA interferases. Furthermore, it is clear that TA systems crosstalk at least partly.
Discussion
Preceding studies have indicated that the primary role of MazF is cleaving bulk mRNA and inhibiting global translation (Zhang et al. 2003; Sauert et al. 2016). This hypothesis seems to be adequate, because only 175 of all 4318 E. coli genes (4.1%) do not contain ACA in their coding sequences (Table 1) (Venturelli et al. 2017). In contrast, RNA-seq analysis in this study indicated that MazF activation leads to biased regulation of RNA levels, which is independent of the presence and number of ACA sequences. Therefore, we conclude that MazF activation does not result in global and nonspecific translational inhibition and causes cells to shift to a specific cellular state. In agreement with this model, preceding studies have demonstrated that MazF activation leads to the selective synthesis of specific proteins (Amitai et al. 2009; Moll and Engelberg-Kulka 2012; Zorzini et al. 2016). It has also been demonstrated that not all ACA sequences in mRNAs are cleaved by MazF in vitro and in vivo (Tripathi et al. 2014), further supporting this model. Nevertheless, we do not exclude the possibility of global inhibition of translation by MazF, because in mazF-overexpressing cells, artificial ACA-free mRNAs are almost exclusively translated into proteins (Suzuki et al. 2006), and because MazF functions as an RNA interferase even in mammalian cells, mediating cellular mRNA degradation, inhibition of protein synthesis, and the induction of apoptosis (Shimazu et al. 2007).
An important question is how cellular levels of individual RNA species are adjusted following MazF activation. One possibility is modulation through RNA decay. As mentioned above, it seems that cleavage at ACA sequences occurs non-uniformly. Although the exact reason for this remains unclear, MazF seems to cleave at ACA sequences only within unstructured RNA regions, and those within structured regions are sterically hindered from cleavage (Zhang et al. 2004; Zhu et al. 2008; Vesper et al. 2011). The fate of cleaved RNA is still unclear, because 3′-ends of MazF-cleaved RNA are phosphate groups; RNAs cleaved by typical processing endonucleases such as RNases E/G bear 3′-hydroxyl groups, and RNases E/G are the primary endonucleases that trigger mRNA decay (Kushner 2002). Even so, one previous study proposed the possibility that RNA cleaved by MazF is further degraded into four nucleotide monophosphates by RNases (Mok et al. 2015). Another study indicated that in response to stress, several RNA interferases might trigger mRNA decay as RNases E/G do (Deutscher 2006). Taken together, we propose a model, wherein following MazF activation, the cellular level of a specific RNA species is adjusted through the non-global cleavage of RNA at ACA sites and the further degradation of the cleaved RNA. However, we have shown here that MazF activation evokes the activation of other RNA interferases (Table 6), and thus, a more complicated cleavage and decay pathway must be considered.
Alternatively, RNA levels after MazF activation can be adjusted at the transcriptional level. However, the expression of all seven sigma factor genes was unchanged in this study (Supplementary Table S1). It should be noted that MazEF functions as a DNA-binding transcriptional repressor, and that MazE or the MazEF complex, alone or in conjunction, repress transcription from its own promoter (Marianovsky et al. 2001). Since, neither MazE nor the MazEF complex, alone or in conjunction, have been reported to regulate transcription from other promoters, we searched for the DNA-binding motif of the MazEF complex (gTATcTAcAATnnanATTGATATATAC, where n is any nucleotide and capital letters denote important nucleotides) (Marianovsky et al. 2001) in the MG1655 chromosome. We failed to find highly homologous sequences except for the 5′-upstream region of the mazEF operon and found sequences showing low similarity in the 5′-upstream region of mntP, yfgG, and tauA (Supplementary Table S7). Among these three genes, only mntP was differentially expressed (log2-fold change = 3.1, FDR = 8.4 × 10–3). In addition, information regarding promoters of upregulated genes in segments I and II were collected, but common features were not found (Supplementary Table S8). Collectively, we could find no obvious evidence of adjustment at the transcriptional level. Therefore, if RNA levels are adjusted at the transcriptional level, this modulation is presumably not governed by a single transcription factor, but rather a complex network.
Note that mechanisms of RNA modulation, both through RNA decay and at the transcriptional level can be compatible. Such mechanisms might work separately depending on the target genes. We believe that how cellular levels of individual RNA species are adjusted following MazF activation is an important target of future research. To unveil the function of MazEF, how indiscriminate cleavage of RNA and how specific adjustment of individual RNA species are associated should be addressed.
The finding that upregulated gene clusters are present in putative horizontally acquired DNA fragments (Fig. 1 and Supplementary Figs S2 and S3) is particularly interesting with respect to bacterial genome evolution. Many bacterial TA systems including E. coli mazEF are thought to enter the bacterial chromosome thorough horizontal gene transfer (Van Melderen and Saavedra De Bast 2009; Ramisetty and Santhosh 2016) and are frequently found on plasmids, prophages, transposons, and integrons (Bustamante et al. 2014; Mruk and Kobayashi 2014). In general, most horizontally transferred genes are lost from chromosomes just after this event (Kunin and Ouzounis 2003), because they are dispensable and metabolic burden is increased to replicate DNA (Cavalier-Smith 2005; Giovannoni et al. 2014). Nevertheless, portions of these elements can be retained on chromosomes, which can confer the ability to utilize specific nutrients or elevate tolerance to stresses. MazF might contribute to the maintenance of horizontally acquired genes by upregulating expression of such genes, so that cells can survive in harsh conditions. Under harsh conditions, MazF might stimulate adaptive evolution and generating phenotypic diversity. As mentioned above, the promoter sequences of upregulated genes in segments I and II have no common features (Supplementary Table S8), and crosstalk of TA systems are observed (Table 6). Therefore, we assume that the gene expression regulation in these segments is not governed by a single mechanism, but are co-ordinated and co-evolved each other.
In conclusion, both the validation (or expanded discovery) of already known facts and novel discoveries are described regarding the cellular role of MazF. The former includes (1) the selective synthesis of specific proteins, (2) alterations in chaperon networks, and (3) crosstalk between TA systems. The latter includes the fact that (1) the presence and number of ACA sequences is not related to cellular RNA levels, (2) upregulated gene clusters are present in putative horizontally acquired DNA fragments, (3) thiamine and amino acid metabolism genes are specifically regulated, and (4) the energy acquisition pathway is limited at multiple steps.
References
Aakre CD, Phung TN, Huang D, Laub MT (2013) A bacterial toxin inhibits DNA replication elongation through a direct interaction with the β sliding clamp. Mol Cell 52:617–628
Aiba H, Adhya S, de Crombrugghe B (1981) Evidence for two functional gal promoters in intact Escherichia coli cells. J Biol Chem 256:11905–11910
Amitai S, Kolodkin-Gal I, Hananya-Meltabashi M, Sacher A (2009) Engelberg-Kulka H (2009) Escherichia coli MazF leads to the simultaneous selective synthesis of both "death proteins" and "survival proteins". PLoS Genet 5:e1000390
Blumer C, Kleefeld A, Lehnen D, Heintz M, Dobrindt U, Nagy G, Michaelis K, Emödy L, Polen T, Rachel R, Wendisch VF, Unden G (2005) Regulation of type 1 fimbriae synthesis and biofilm formation by the transcriptional regulator LrhA of Escherichia coli. Microbiology 151:3287–3298
Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120
Bustamante P, Tello M, Orellana O (2014) Toxin–antitoxin systems in the mobile genome of Acidithiobacillus ferrooxidans. PLoS ONE 9:e112226
Cavalier-Smith T (2005) Economy, speed and size matter: evolutionary forces driving nuclear genome miniaturization and expansion. Ann Bot 95:147–175
Culviner PH, Laub MT (2018) Global analysis of the E. coli toxin MazF reveals widespread cleavage of mRNA and the inhibition of rRNA maturation and ribosome biogenesis. Mol Cell 70:868–880
Deutscher MP (2006) Degradation of RNA in bacteria: comparison of mRNA and stable RNA. Nucleic Acids Res 34:659–666
Ellrott K, Yang C, Sladek FM, Jiang T (2002) Identifying transcription factor binding sites through Markov chain optimizations. Bioinformatics 18(Suppl 2):S100–S109
Ferenci T, Zhou Z, Betteridge T, Ren Y, Liu Y, Feng L, Reeves PR, Wang L (2009) Genomic sequencing reveals regulatory mutations and recombinational events in the widely used MC4100 lineage of Escherichia coli K-12. J Bacteriol 191:4025–4029
Giovannoni SJ, Thrash JC, Temperton B (2014) Implications of streamlining theory for microbial ecology. ISME J 8:1553–1565
Huang DW, Sherman BT, Lempicki RA (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Kanehisa M, Goto S, Sato Y, Furumichi M, Tanabe M (2012) KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res 40(Database issue):D109–D114
Karcagi I, Draskovits G, Umenhoffer K, Fekete G, Kovács K, Méhi O, Balikó G, Szappanos B, Györfy Z, Fehér T, Bogos B, Blattner FR, Pál C, Pósfai G, Papp B (2016) Indispensability of horizontally transferred genes and its impact on bacterial genome streamlining. Mol Biol Evol 33:1257–1269
Kasari V, Mets T, Tenson T, Kaldalu N (2013) Transcriptional cross-activation between toxin–antitoxin systems of Escherichia coli. BMC Microbiol 13:45
Kim Y, Wang X, Ma Q, Zhang XS, Wood TK (2009) Toxin–antitoxin systems in Escherichia coli influence biofilm formation through YjgK (TabA) and fimbriae. J Bacteriol 191:1258–1267
Kim Y, Wang X, Zhang XS, Grigoriu S, Page R, Peti W, Wood TK (2010) Escherichia coli toxin/antitoxin pair MqsR/MqsA regulate toxin CspD. Environ Microbiol 12:1105–1121
Kolodkin-Gal I, Verdiger R, Shlosberg-Fedida A, Engelberg-Kulka H (2009) A differential effect of E. coli toxin–antitoxin systems on cell death in liquid media and biofilm formation. PLoS ONE 4:e6785
Kunin V, Ouzounis CA (2003) The balance of driving forces during genome evolution in prokaryotes. Genome Res 13:1589–1594
Kushner SR (2002) mRNA decay in Escherichia coli comes of age. J Bacteriol 184:4658–4665
Magoc T, Wood D, Salzberg SL (2013) EDGE-pro: estimated degree of gene expression in prokaryotic genomes. Evol Bioinform Online 9:127–136
Marianovsky I, Aizenman E, Engelberg-Kulka H, Glaser G (2001) The regulation of the Escherichia coli mazEF promoter involves an unusual alternating palindrome. J Biol Chem 276:5975–5984
Masuda H, Tan Q, Awano N, Wu KP, Inouye M (2012) YeeU enhances the bundling of cytoskeletal polymers of MreB and FtsZ, antagonizing the CbtA (YeeV) toxicity in Escherichia coli. Mol Microbiol 84:979–989
McCarthy JD, Chen Y, Smyth GK (2012) Differential expression analysis of multifactor RNA-seq experiments with respect to biological variation. Nucleic Acids Res 40:4288–4297
Mets T, Kasvandik S, Saarma M, Maiväli Ü, Tenson T, Kaldalu N (2019) Fragmentation of Escherichia coli mRNA by MazF and MqsR. Biochimie 156:79–91
Mok WW, Park JO, Rabinowitz JD, Brynildsen MP (2015) RNA futile cycling in model persisters derived from MazF accumulation. mBio 6:e01588-15
Moll I, Engelberg-Kulka H (2012) Selective translation during stress in Escherichia coli. Trends Biochem Sci 37:493–498
Mruk I, Kobayashi I (2014) To be or not to be: regulation of restriction–modification systems and other toxin–antitoxin systems. Nucleic Acids Res 42:70–86
Nonaka G, Blankschien M, Herman C, Gross CA, Rhodius VA (2006) Regulon and promoter analysis of the E. coli heat-shock factor, sigma32, reveals a multifaceted cellular response to heat stress. Genes Dev 20:1776–1789
Ramisetty BC, Santhosh RS (2016) Horizontal gene transfer of chromosomal Type II toxin–antitoxin systems of Escherichia coli. FEMS Microbiol Lett 363:fnv238
Sauert M, Wolfinger MT, Vesper O, Müller C, Byrgazov K, Moll I (2016) The MazF-regulon: a toolbox for the post-transcriptional stress response in Escherichia coli. Nucleic Acids Res 44:6660–6675
Shimazu T, Degenhardt K, Nur-E-Kamal A, Zhang J, Yoshida T, Zhang Y, Mathew R, White E, Inouye M (2007) NBK/BIK antagonizes MCL-1 and BCL-XL and activates BAK-mediated apoptosis in response to protein synthesis inhibition. Genes Dev 21:929–941
Stothard P, Wishart DS (2005) Circular genome visualization and exploration using CGView. Bioinformatics 21:537–539
Suzuki M, Roy R, Zheng H, Woychik N, Inouye M (2006) Bacterial bioreactors for high yield production of recombinant protein. J Biol Chem 281:37559–37565
Tripathi A, Dewan PC, Siddique SA, Varadarajan R (2014) MazF-induced growth inhibition and persister generation in Escherichia coli. J Biol Chem 289:4191–4205
Van Melderen L, Saavedra De Bast M (2009) Bacterial toxin–antitoxin systems: more than selfish entities? PLoS Genet 5:e1000437
Venturelli OS, Tei M, Bauer S, Chan LJG, Petzold CJ, Arkin AP (2017) Programming mRNA decay to modulate synthetic circuit resource allocation. Nat Commun 8:15128
Vesper O, Amitai S, Belitsky M, Byrgazov K, Kaberdina AC, Engelberg-Kulka H, Moll I (2011) Selective translation of leaderless mRNAs by specialized ribosomes generated by MazF in Escherichia coli. Cell 147:147–157
Wang X, Kim Y, Ma Q, Hong SH, Pokusaeva K, Sturino JM, Wood TK (2010) Cryptic prophages help bacteria cope with adverse environments. Nat Commun 1:147
Wang X, Lord DM, Cheng HY, Osbourne DO, Hong SH, Sanchez-Torres V, Quiroga C, Zheng K, Herrmann T, Peti W, Benedik MJ, Page R, Wood TK (2012) A new type V toxin–antitoxin system where mRNA for toxin GhoT is cleaved by antitoxin GhoS. Nat Chem Biol 8:855–861
Yamaguchi Y, Inouye M (2011) Regulation of growth and death in Escherichia coli by toxin–antitoxin systems. Nat Rev Microbiol 19:779–790
Yamazaki Y, Niki Y, Kato J (2008) Profiling of Escherichia coli chromosome database. Methods Mol Biol 416:385–389
Zhang Y, Zhang J, Hoeflich KP, Ikura M, Qing G, Inouye M (2003) MazF cleaves cellular mRNAs specifically at ACA to block protein synthesis in Escherichia coli. Mol Cell 12:913–923
Zhang J, Zhang Y, Zhu L, Suzuki M, Inouye M (2004) Interference of mRNA function by sequence-specific endoribonuclease PemK. J Biol Chem 279:20678–20684
Zhu L, Phadtare S, Nariya H, Ouyang M, Husson RN, Inouye M (2008) The mRNA interferases, MazF-mt3 and MazF-mt7 from Mycobacterium tuberculosis target unique pentad sequences in single-stranded RNA. Mol Microbiol 69:559–569
Zorzini V, Mernik A, Lah J, Sterckx YG, De Jonge N, Garcia-Pino A, De Greve H, Versées W, Loris R (2016) Substrate recognition and activity regulation of the Escherichia coli mRNA endonuclease MazF. J Biol Chem 291:10950–10960
Acknowledgements
We would like to thank to the rest of iGEM team Tokyo_Tech 2016 team members for collaboration on the early stages of this work. We are grateful to Ms. Tomoko Narisawa and Ms. Masako Sato for technical assistance. This work was supported by the Japan Society for the Promotion of Science (Grant number 17K07714).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing financial conflicts of interests.
Additional information
Communicated by Erko Stackebrandt.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Akiyama, K., Fujisawa, K., Kondo, H. et al. MazF activation causes ACA sequence-independent and selective alterations in RNA levels in Escherichia coli. Arch Microbiol 202, 105–114 (2020). https://doi.org/10.1007/s00203-019-01726-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00203-019-01726-9