
Abstract
Allergic asthma is an inflammatory disease of the airways characterized by recurrent episodes of wheezing and bronchoconstriction. Chronic inflammation may finally lead to structural damage followed by airway remodeling. Various studies in recent years contributed to unravel important aspects of the immunopathogenesis of asthma and adapted new pharmaceutical developments. Here, I consider some novel insights into the immunopathogenesis of asthma and the protective and pathogenic roles of some innate and adaptive immune cells as well as the function of soluble mediators such as cytokines. Particular attention will be given to new concepts on resolution of chronic airway inflammation for prevention of airway structural damage.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
More than 300 million people worldwide suffer from asthma, a chronic inflammatory disorder of the airways characterized by local inflammation and airway hyperresponsiveness [1]. Possible symptoms include wheezing, coughing, chest tightness, and acute exacerbation associated with breathlessness. It has been calculated that asthma leads to more than 300,000 hospital discharges a year and considerable national annual healthcare resource use (11.0 million physician office visits, 1.7 million emergency room visits, and 1.3 million hospital outpatient department visits according to the Centers for Disease Control and Prevention) in the USA. Several studies suggested a major risk to reappearance of asthma in elderly if one was affected as a child. Allergic asthma may cause debilitating daily symptoms and may be complicated by acute exacerbations of symptoms. Viral infections are common triggers of exacerbations in asthma and may impair asthma symptom control [2, 3]. In addition to viral infections, bacterial infections of the respiratory tract have been associated with worsening or increased severity of symptoms in asthma patients [4] highlighting the concept that various infections may trigger disease exacerbations. It should be noted that asthma is characterized by a marked heterogeneity in pathophysiology and etiology in individual patients. This heterogeneity is mirrored by different disease-inducing mechanisms (allergic versus non-allergic forms of asthma), differences in immune cells acting as key drivers of disease activity (eosinophilic versus non-eosinophilic/neutrophilic forms of asthma), and varieties in both the severity (mild, moderate versus severe forms) and frequency of clinical disease manifestations.
In addition to intrinsic asthma, exogenously triggered forms of asthma exist that can be induced by exposure to allergens [5,6,7,8,9,10]. Allergic asthma is a consequence of complex gene–environment interactions and flares are frequently triggered by allergen exposure. The recommendation for the resolution of acute symptoms of this disease (e.g., wheezing, coughing, breathlessness) is to avoid contact with allergen exposure in the environment. In addition, a reduction of fat- or sugar-rich diet may be helpful. Furthermore, therapeutic asthma strategies comprise anti-inflammatory and bronchodilator treatments. In particular, inhaled corticosteroids, alone or in combination with long-acting bronchodilators, are frequently used for asthma therapy. However, symptom minimization may not be achieved in some patients leading to the manifestation of uncontrolled asthma. As specified below, asthma therapy has changed in recent years through the introduction of biological agents such as monoclonal antibodies. These new biological therapies for asthma, together with developments in the field of serum biomarkers, are important new opportunities for phenotype-specific interventions and personalized medicine in the field of asthma. The specific immunological molecules targeted by monoclonal antibodies have been identified based on the characterization of the cellular and molecular components present in the airways of asthmatic patients. In this regard, it has been recognized in the last 30 years that the airways of many asthmatic patients are filled with inflammatory cells like eosinophils during exacerbations. Thus, many biological therapies have been designed to target eosinophil activation or function in patients with asthma.
Soluble mediators like cytokines produced by immune cells have been shown to play a fundamental role in the pathophysiology of asthma. For instance, genome-wide differential gene expression in response to dust mite allergen identified cytokines such as IL-9 as gene targets that might interact with environmental dust mite to increase severe asthma exacerbations in children [11]. Moreover, pathways of innate immune stimulation by environmental or endogenous pathogen-associated molecular patterns (PAMPs) and danger-associated molecular patterns (DAMPs) play a key role in asthma pathogenesis [12]. Specifically, viral and microbial stimuli as well as factors of the airway microbiota can trigger pattern recognition receptors and may contribute to the shaping of the inflammatory response seen in asthma [4]. Activated PAMPs and DAMPs have been demonstrated to modulate mucosal immune responses of the airways via effects on cell activation and trafficking. In particular, mucosal immune cells such as dendritic cells, macrophages, granulocytes, innate lymphoid cells (ILCs), B cells, and T lymphocytes can be directly or indirectly activated via environmental triggers. Subsequently, these immune cells produce proinflammatory mediators that drive and perpetuate mucosal inflammation and airway remodeling. Cytokines of the Th2 group such as IL-4, IL-5, and IL-13 have a key role in amplifying mucosal inflammation. However, studies in recent years have clearly demonstrated that only a subgroup of patients with asthma has elevated Th2 type cytokines. In fact, only approximately 50% of asthma patients demonstrate high type 2 cytokine responses and/or elevated numbers of eosinophils. The other group of patients (“type 2-low”) is frequently characterized by a different immune phenotype associated with airway neutrophilia, goblet cell hyperplasia, and obesity-related systemic inflammation [13,14,15,16,17]. These findings highlight the concept that several asthma phenotypes exist in clinical practice and may require individual therapeutic approaches for optimized therapy.
Immune cells and signaling pathways in asthma
The chronic inflammation in allergic asthma is associated with marked airway hyperresponsiveness and leads to recurrent episodes of exacerbation marked by wheezing, breathlessness, chest tightness, and coughing [18]. Studies in recent decades have unequivocally shown that immune cell pathways play a crucial role in the pathogenesis of asthma. For instance, immunoglobulin E (IgE) bound to the surface of immune cells such as mast cells may bind to specific airway allergens followed by IgE cross-linking, cell activation, and preformed mediator release [19,20,21]. Mediators include histamine, a potent proinflammatory substance leading to vasodilatation and local inflammation. In addition to mast cells, other mucosal immune cells in the airways such as innate lymphoid cells (ILCs), macrophages, dendritic cells, and adaptive immune cells such as B and T lymphocytes play an important role in the pathogenesis of asthma. In particular, these cell types have been shown to produce soluble mediators such as cytokines that impair or boost mucosal inflammation of the airways in allergic asthma. Moreover, these cell types may respond to cytokine signals in the local environment triggering immune cell activation and further cytokine production. For instance, eosinophils have been classically described as proinflammatory immune cells in asthma and these cells differentiate from progenitor cells derived from the bone marrow under the influence of cytokines such as IL-3, IL-5, and GM-CSF [13, 22, 23]. In addition, IL-3 has been demonstrated to contribute to the differentiation of mucosal bone marrow–derived mast cells and to basophil differentiation and activation [24]. Remarkably, these two cell types are not the predominant cells in the airways during asthma exacerbations, but they may contribute more than other inflammatory cells to the terminal exacerbation of the allergic reaction. In fact, these cell types carry the high affinity IgE receptor on their surface [18, 25], and after allergen encounter cross-link, the allergen on the IgE bound to the high affinity IgE receptor resulting in the release of potent preformed mediators like histamine that favor bronchoconstriction. In addition to histamine, mast cells in asthma may produce various other mediators including chymase, tryptase, prostaglandin D2, and leukotriene C4, and thus favor the induction of mucosal edema, bronchoconstriction, muscle hyperplasia and dysfunction, and mucin production through multiple pathways. The number of mast cells is significantly increased in patients with asthma. These cells are located in multiple areas within the airways including the epithelium, the submucosal glands, and the smooth muscle layers. In addition to the above mediators, mast cells have been demonstrated to produce numerous proinflammatory or pro-fibrotic cytokines including IL-4, IL-13, IL-33, TGFβ1, and TSLP. These cytokines have been suggesting to control various key features of the pathophysiology of asthma including epithelial cell shedding, goblet cell and muscle cell hyperplasia, hypervascularization, augmented collagen production, and subepithelial fibrosis [21]. Thus, mast cells emerge as key players in the pathophysiology of asthma and targeting of mast cell activation and function appears of high relevance to achieve resolution of disease.
One of the biologicals targeting activation of IgE receptor–bearing immune cells such as mast cells consists of omalizumab which is a humanized monoclonal antibody that specifically binds to free human immunoglobulin E (IgE) to affect the inflammatory process caused by mast cell activation in patients with moderate-to-severe allergic asthma [5, 26]. This therapeutic approach is like many other currently used anti-inflammatory medications positioned downstream of the allergic reaction in asthma [27]. Looking at other biological targets, it has been reasoned that eosinophils represent the predominant cell type populating the airways of patients during asthma exacerbations in many patients and they may contribute with their cytotoxic content to the destruction of the airway epithelial barrier, thus resulting in major chronic airway disorder that is difficult to reverse.
There is increasing evidence that eosinophils may not only mediate Th2 responses but also potently regulate homeostatic processes at steady state, thus challenging the paradigm of eosinophils as a merely destructive and inflammatory cell type [28]. Under baseline conditions, eosinophils rapidly leave the bloodstream to enter mucosal tissues, like gut and lung, where they regulate important immune functions. These so-called resident eosinophils (rEos) have been recently demonstrated to be morphologically and phenotypically distinct from the inflammatory eosinophils (iEos) that are recruited to the lung during house dust mite (HDM)–induced allergic inflammation [29]. Moreover, rEos have been shown to prevent the development of pulmonary Th2 immunity, and thus contribute to lung homeostasis. These eosinophils have a ring-shaped nucleus and reside in the parenchyma as opposed to those iEos increased in asthma which are induced in the bronchial lumen and have a segmented nucleus. The relationship between rEos and iEos is not entirely clear; however, further investigation is needed. Some reports indicated that rEos are IL-5 independent [30] and suggested that rEos promote the development of Th1 immunity, either by directly inhibiting T cells or by impairing the ability of dendritic cells to induce Th2 immunity [29]. The discovery of this new type of eosinophils raises potential concern about therapies trying to block eosinophil function in eosinophilic asthma, as such, therapies might also affect the function of rEos, and thus tissue homeostasis. Also, considering asthma exacerbations driven by respiratory viruses like rhinovirus, eosinophils play an important role in clearing the infections. These functions of eosinophils are not well examined yet but their role in infections must be further investigated during the resolution of asthma (Fig 1).
Neutrophils have been suggested to play an important pathogenic role in asthma patients with a “type 2-low” immune response. Accordingly, various studies have addressed the contribution of neutrophils to asthma pathogenesis in recent years [31,32,33,34]. It has been suggested that tissue neutrophils in the lung may possess specific phenotypic features distinguishing them from resting blood neutrophils. Moreover, neutrophil-derived mediators and CXCR4+-derived neutrophil extracellular traps have been suggested to amplify the underlying allergic immune response and cardinal features of allergic asthma [35]. However, further studies are needed to gain further insights into the role of neutrophils in allergic asthma.
Finally, macrophages have been implicated in asthma pathogenesis [36,37,38,39]. Monocytes recruited into mucosal tissues can interact with environmental factors leading to macrophage polarization. In this context, macrophages can differentiate in various different subsets like M1-like, classically activated or M2-like alternatively activated cells. On exposure to local microenvironments, recruited macrophages can be polarized into either classically activated or alternatively activated phenotypes. Activated macrophages have been suggested as potential indicators of oxidative stress, tissue remodeling, and disease severity in asthma [37]. Moreover, studies in macrophage-deficient mice suggested an important role of these cells in the asthmatic phenotype. Specifically, studies in CD163-deficient mice showed a marked reduction of proinflammatory cytokines in the bronchioalveolar lavage fluid when compared with control wild-type mice [38]. These studies suggested that macrophages may play a key role in asthma and should be considered as targets for new therapeutic approaches.
Targeting Th2 and Th9 cytokines for resolution of asthma
Th2 and Th9 cells and their signature cytokines have been shown to play a key role in asthma [40] [41, 42]. Th2 cells are characterized by production of IL-4, IL-5, IL-6, and IL-13. In addition to Th2 T cells, these cytokines can be produced by innate lymphoid type 2 cells in asthma [43]. Th2 cytokines or their receptors have been targeted in patients with asthma or murine models of disease in order to achieve resolution of asthma. In particular, studies targeting IL-13 were very successful to block allergic airway inflammation and airway hyperresponsiveness in experimental asthma. In addition to targeting single cytokines or cytokine receptors, transcription factors have been emerged as possible targets for blockade of several cytokines simultaneously. One example consists of GATA-3. Increased GATA-3 expression was noted in asthmatic airways and correlated with the presence of IL-5 mRNA+ cells. These results suggested a causal association between augmented GATA-3 expression and dysregulated IL-5 expression in atopic asthma [44]. Treatment with GATA-3 antisense DNA was effective in suppressing airway inflammation and hyperresponsiveness in experimental asthma [45]. Additionally, GATA-3 inhibition in asthma patients resulted in multiple downstream therapeutical effects including suppression of airways eosinophilia and mucus production.The relevance of GATA-3 for Th2 cytokine production was underlined by biomarker analysis in this study, as biomarkers indicated an attenuation of Th2-regulated inflammatory responses upon GATA-3 blockade [46]. Other studies attempted to target the transcription factor BATF or the Janus kinase Tyk-2 to block IL-4 and IL-9 pathways, and these studies demonstrated resolution of allergic experimental asthma upon blockade of BATF or Tyk-2, respectively [40, 47].

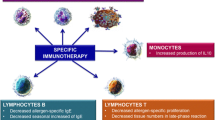
Resolution of allergic asthma. Allergic asthma exacerbations are associated with increased accumulation of Th2 in the airways which release TH2 (Th2) cytokines like IL-5, IL-9, and IL-13. These cytokines recruit inflammatory cells belonging to the innate immunity. Usually, the immune asthmatic reaction is reversible and this resolution is associated with decreased inflammatory cells like Th2 cells, mast cells, basophils, inflammatory eosinophils (iEos), and induction of resident eosinophils rEos and TH1 (Th1) and Tregulatory cells producing IL-10 (TR1=Tr1) or TGF-beta and IL-10. Chronic asthma is a condition in which the Th2-driven inflammation does not resolve and results in a profound airway remodeling dominated by pro-fibrotic cytokines like TGF-beta
Th9 cells are a specific subgroup of activated lymphocytes characterized by the production of IL-9 [48,49,50,51,52]. These cells are associated with the expression of the transcription factor PU.1 [50, 53]. In mice, increased levels of IL-9 in the airways of naive animals induced lung eosinophilia and serum total IgE levels, which are 2 clinical features of asthma. These data supported a central role for the IL-9 pathway in the pathogenesis of allergic inflammation [54]. Furthermore, p-STAT6 and PU.1 were associated with Th9 cells and IL-9 production in allergen-induced airway inflammation [55]. In humans, genetic studies suggested a link between the IL-9R locus and asthma in patients. Studies in humans further revealed that patients with allergic asthma have increased peripheral blood Th9 cell counts and serum IL-9, while eosinophil apoptosis was inversely related to IL-9 concentration [56]. B lymphocyte-induced maturation protein 1 (Blimp-1), a negative regulator of Th9 development, appeared to orchestrate the resolution of airway inflammation in patients with allergic asthma [57]. These findings indicated that IL-9 is specifically upregulated after local allergen challenge in the lungs of atopic asthmatic patients. The increased expression and its correlation with eosinophil numbers suggested a potential role for IL-9 in the late phase of the allergic response [58]. Moreover, IL-9R was expressed on neutrophils from patients with asthma and additional studies demonstrated that IL-9 stimulation results in production of IL-8 [59] highlighting a potential role of IL-9 for mucosal inflammation in asthma. Additionally, IFN-gamma stimulated airway epithelial cells expressed IL-9R suggesting that IL-9 signaling may affect mucosal immune regulation via several independent pathways [60]. Moreover, anti-IL-9 antibody treatment suppressed production of IL-4, IL-5, and IL-13 in bronchoalveolar lavage fluid suggesting that blockade of IL-9 might be useful for treatment of asthma [61]. Finally, additional studies revealed that IL-9 levels are highly correlated to expression levels of the calcium-activated chloride channel HCLCA1 and mucus production of airway epithelium in asthma suggesting that this cytokine is a potential marker for atopic asthma [62, 63]. However, a clinical trial using a neutralizing IL-9 antibody (MEDI-528) to existing asthma controller medications showed no improvement in asthma exacerbation rates or FEV1 values [64].
Interferons and viral infections in asthma
Common exacerbations of asthma especially in children have been associated with the detection of rhinovirus in the airways. Rhinovirus, a member of the picornaviridae family, is often detected in the airways of asthmatic children during symptomatic visits to the hospital because of an ongoing infection in the airways [65]. The immune responses to rhinovirus comprise the upregulation of interferon pathways [66]. Especially type I and type III interferons were found to be associated with rhinovirus infection of the airways [67]. Dysregulated interferon responses have been observed in asthmatic patients during rhinovirus-induced exacerbations in non-controlled asthmatics [68]. During acute rhinovirus infection, IFN type I was found to be induced in serum of the cohort of preschool children with asthma in symptomatic visits. In vitro exposure of peripheral blood mononuclear cells (PBMCs) from asthmatic and control children to rhinovirus demonstrated in PBMCs of asthmatic children RV-induced upregulation of genes of the IFN pathway like STAT1/2, IRF1, but also programmed cell death protein 1 ligand (PDL1), a gene induced by IFN but implicated in T cell exhaustion [66, 69] and cytotoxic T lymphocyte-associated protein 4 (CTLA4), a regulatory protein that downregulates mucosal immune responses. Thus, viral infections such as those induced by rhinoviruses lead to complex alterations of mucosal immune responses in the airways with particular changes in IFN regulation. In summary, our findings in preschool children with asthma indicate that their immune system is still able to react during acute exacerbation of disease with IFN type I and III immune responses [66]. However, this reaction remains associated with induced immune Inhibitory pathways like PDL1 and CTLA4 that need further attention. Similarly, we found that during acute rhinovirus infections, endogenous TGF-β is retained intracellularly in rhinovirus-infected cells, resulting in a T-bet–mediated immune response. However, rhinovirus infection also activates TGF-β present in the environment, as in patients with chronic asthma, to replicate and inhibit effective antiviral immune responses. Thus, it is possible that children with acute asthma are able to induce an effective anti-rhinovirus immune response during acute exacerbation. By contrast, in patients with chronic asthma, active TGF-β is induced and released by structural cells. It can be that in chronic asthma rhinovirus induces the effect of the exogenous TGF-β resulting in TH1 and TC1 depletion. These data open new avenues for our understanding of the role of rhinovirus-mediated asthma in children (Fig 1).
Th1 and Th17 cytokines and cytokines produced by regulatory T cells in asthma
Th1 T lymphocytes are characterized by the production of IFN-gamma and other immunoregulatory cytokines such as TNF. These T cells develop via IL-12 stimulation and express the transcription factors STAT1, STAT4, and T-bet [70,71,72,73]. Initial studies in patients with asthma detected a marked suppression of T-bet levels suggesting a regulatory role of this transcription factor in asthma pathogenesis [74]. Consistently, mice deficient in T-bet displayed some features of asthma upon OVA sensitization including airway inflammation and hyperresponsiveness underlining a key role of T-bet in airway inflammation [74]. Additional studies indicated that modulation of Th1 responses may affect lung immunology and experimental asthma. In fact, it was found that preventive intranasal administration of IL-27 induces a Th1 environment in the lung that helps to alleviate Th2-mediated allergic asthma by boosting the STAT1 rather than the STAT4 pathway [75].
Th17 cells produce various immunoregulatory cytokines such as IL-17A, IL-17F, IL-21, and IL-22 [76, 77]. These cells are characterized by the expression of the transcription factor RORgamma-t. Several studies implicated a role of Th17 cells in asthma pathogenesis [78,79,80,81,82]. In particular, elevated production of IL-17 was found in a subset of patients with severe asthma exhibiting a “type 2-low” phenotype. It has been suggested that the differentiation of IL-17-producing cells is favored in severe asthma via a micromilieu rich in IL-6 and TGF-beta. Particularly high levels of IL-17 were noted in geriatric asthma. In fact, a study in geriatric asthma showed that serum IgE, IL-17A, IL-17F, and GR-beta levels were significantly elevated in patients with moderate or severe asthma as compared to young patients with moderate asthma and control patients [83]. Asthma patients with high levels of IL-17 displayed mainly features of neutrophilic asthma with goblet cell and smooth muscle hyperplasia, high mucus production, and airway inflammation dominated by neutrophils. As airway epithelial cells and smooth muscle cells may express IL-17R, these findings suggested that IL-17 may drive airway remodeling in asthma. Although pleiotropic roles of IL-17 were noted in experimental asthma in mice, the above findings suggested that IL-17 may play an important role in airway pathophysiology. Finally, it was found that Th17 cytokines are associated with steroid resistance in asthma suggesting that these cytokines may play an active role in molecular resistance against therapy.
The balance between Th17 cells and regulatory T cells (Treg cells) has been suggested to have a key role in the pathogenesis of asthma [78,79,80,81,82]. Various studies indicated that the homeostatic balance between Treg and Th17 cells is markedly altered in asthma exacerbation and correlates with asthma severity [84]. Regulatory T cells are known to produce immunosuppressive cytokines such as IL-10 and TGF-beta [69, 85, 86]. These cytokines may suppress proinflammatory mucosal immune responses in asthma [87]. For instance, in a murine model of asthma induced by repeated house dust mite inhalation, a mixed IL-13/IL-17A cytokine response and an accumulation of IL-10-producing T cells was noted in the lungs. Ablation of T cell-derived IL-10 boosted IFN-gamma and IL-17A cytokine responses in experimental asthma suggesting that IL-10 from effector T cells signals to CD11c+ myeloid cells to suppress a pathogenic cytokine response in asthma [88]. In patients with severe asthma, suppression of IL-10 levels was noted indicating an impaired capacity of the lung immune system to counteract proinflammatory signals [89]. Although IL-10 and TGF-beta may suppress inflammation, they may also cause structural alterations, as TGF-beta has been identified as pro-fibrotic cytokine that markedly affects lung fibrosis and airway remodeling in asthma [87, 90, 91].
Treatment of patients with asthma: targeting of resolution via immunotherapy
Allergen-specific immunotherapy represents a possibly effective treatment for allergic asthma in experimental model systems and patients with active disease [92]. This therapeutic approach is based on repeated administration of small amounts of the sensitizing allergen. Experimental studies have indicated that allergen immunotherapy induces IL-10 production and antigen-specific Foxp3+ Treg cells and that such Treg cells control and orchestrate the anti-inflammatory pathways driving resolution of inflammation [93]. Meta-analyses of clinical studies suggested that allergen-specific immunotherapy may lead to reductions in symptom and medication scores. However, no clear, consistent evidence was obtained that this therapeutic approach improves measures of lung function [94, 95].
In addition to allergen immunotherapy, additional immunotherapies for allergic asthma exist that are based on suppression of IgE or mucosal cytokine responses. For instance, omalizumab, a recombinant humanized IgG1 monoclonal antibody, was approved in the USA as an add-on treatment in moderate-to-severe allergic asthma and in Europe for severe allergic asthma. This antibody binds to the Fc region of IgE [96, 97]. Thereby, omalizumab binds to circulating IgE and forms biologically inert IgE–anti-IgE complexes which do not activate the complement cascade. In clinical trials, omalizumab reduced both early- and late-phase asthmatic responses after allergen inhalation challenge and suppressed the numbers of eosinophils in sputum as well as FcεRI expression on various immune cell subsets. It has been suggested that the reduction in the expression of FcεRI on dendritic cells may impair allergen processing in asthma [5, 98, 99]. Numerous clinical studies now underline the long-term effectiveness of omalizumab for reduction of asthma exacerbations and improvement of lung function [96, 97].
Several additional antibodies have been tested in clinical trials for asthma: mepolizumab, reslizumab, and benralizumab [100] [7, 101] [102, 103]. These antibodies target the function of IL-5 and IL-5 signaling in eosinophilic asthma. Eosinophil differentiation, survival, and activation are regulated by IL-5, a cytokine whose receptor is present on the cell surface of eosinophils or basophils. The monoclonal antibody benralizumab binds specifically to the IL-5R and thereby blocks IL-5 signaling and subsequent eosinophil differentiation. Furthermore, benralizumab binds to the RIIIa region of the Fcgamma receptor on immune cells followed by induction of cytotoxicity in eosinophils. By taking advantage of these two independent pathways, benralizumab has been shown to cause effective depletion of eosinophils. The benralizumab-induced depletion of eosinophils is more pronounced as compared to that induced by IL-5 blockers such as mepolizumab and reslizumab [7, 101]. Recent 2 years of safety studies reported no major safety concern upon long-term eosinophil depletion [104]. In addition to antibodies targeting IL-5 or IL-5R in asthma, recent studies also addressed the effects of dupilumab in asthma. This antibody blocks the alpha subunit of the IL-4 and IL-13 receptors, and thus has a dual mechanism of action by inhibiting the activity of two cytokines simultaneously. Clinical trials suggested that the addition of dupilumab is associated with a reduced risk of severe asthma exacerbations and improvement in FEV1 in patients with uncontrolled asthma [105,106,107,108,109,110]. Thus, there are many new current and future options for immunotherapy in asthma that may allow novel approaches for individualized therapy in subgroups of the disease.
Summary
Studies in recent years have highlighted a crucial role of immunotherapy in asthma. In some cases, such immunotherapy has led to complete resolution of the asthmatic phenotype. The discovery of subgroups of immune phenotypes in asthma, however, suggests that personalized medicine is needed to achieve symptom control and improvement of lung function in larger groups of patients. Future studies are needed to identify novel targets for immunotherapy that control resolution of the disease.
References
Hartert TV, Peebles RS Jr (2000) Epidemiology of asthma: the year in review. Curr Opin Pulm Med 6(1):4–9
Xepapadaki P, Bachert C, Finotto S, Jartti T, Konstantinou GN, Kiefer A, Kowalski M, Lewandowska-Polak A, Lukkarinen H, Roumpedaki E, Sobanska A, Sintobin I, Vuorinen T, Zhang N, Zimmermann T, Papadopoulos NG (2018) Contribution of repeated infections in asthma persistence from preschool to school age: design and characteristics of the PreDicta cohort. Pediatr Allergy Immunol 29(4):383–393
Megremis S, Niespodziana K, Cabauatan C, Xepapadaki P, Kowalski ML, Jartti T, Bachert C, Finotto S, West P, Stamataki S, Lewandowska-Polak A, Lukkarinen H, Zhang N, Zimmermann T, Stolz F, Neubauer A, Akdis M, Andreakos E, Valenta R, Papadopoulos NG (2018) Rhinovirus species-specific antibodies differentially reflect clinical outcomes in health and asthma. Am J Respir Crit Care Med
Earl CS, An SQ, Ryan RP (2015) The changing face of asthma and its relation with microbes. Trends Microbiol 23(7):408–418
Strunk RC, Bloomberg GR (2006) Omalizumab for asthma. N Engl J Med 354(25):2689–2695
Chen Q, Guo X, Deng N, Liu L, Chen S, Wang A, Li R, Huang Y, Ding X, Yu H, Hu S, Nie H (2019) alpha-galactosylceramide treatment before allergen sensitization promotes iNKT cell-mediated induction of Treg cells, preventing Th2 cell responses in murine asthma. J Biol Chem 294(14):5438–5455
Chia YL, Yan L, Yu B, Wang B, Barker P, Goldman M, Roskos L (2019) Relationship Between benralizumab exposure and efficacy for patients with severe eosinophilic asthma. Clin Pharmacol Ther 106(2):383–390
Dharmage SC, Perret JL, Custovic A (2019) Epidemiology of asthma in children and adults. Front Pediatr 7:246
Fishe JN, Palmer E, Finlay E, Smotherman C, Gautam S, Hendry P, Hendeles L (2019) A statewide study of the epidemiology of emergency medical services’ management of pediatric asthma. Pediatr Emerg Care
Morgan BW, Grigsby MR, Siddharthan T, Chowdhury M, Rubinstein A, Gutierrez L, Irazola V, Miranda JJ, Bernabe-Ortiz A, Alam D, Wise RA, Checkley W (2019) Epidemiology and risk factors of asthma-chronic obstructive pulmonary disease overlap in low- and middle-income countries. J Allergy Clin Immunol 143(4):1598–1606
Sordillo JE, Kelly R, Bunyavanich S, McGeachie M, Qiu W, Croteau-Chonka DC, Soto-Quiros M, Avila L, Celedon JC, Brehm JM, Weiss ST, Gold DR, Litonjua AA (2015) Genome-wide expression profiles identify potential targets for gene-environment interactions in asthma severity. J Allergy Clin Immunol 136(4):885–92 e2
Holgate ST (2012) Innate and adaptive immune responses in asthma. Nat Med 18(5):673–683
Lambrecht BN, Hammad H, Fahy JV (2019) The cytokines of asthma. Immunity 50(4):975–991
Alagha K, Bourdin A, Vernisse C, Garulli C, Tummino C, Charriot J, Vachier I, Suehs C, Chanez P, Gras D (2019) Goblet cell hyperplasia as a feature of neutrophilic asthma. Clin Exp Allergy 49(6):781–788
Bullone M, Carriero V, Bertolini F, Folino A, Mannelli A, Di Stefano A, Gnemmi I, Torchio R, Ricciardolo FLM (2019) Elevated serum IgE, OCS-dependence and IL-17/22 expression in highly neutrophilic asthma. Eur Respir J
Kalchiem-Dekel O, Yao X, Levine SJ (2019) Meeting the challenge of identifying new treatments for type 2-low neutrophilic asthma. Chest
Ravi A, Chowdhury S, Dijkhuis A, Bonta PI, Sterk PJ, Lutter R (2019) Neutrophilic inflammation in asthma and defective epithelial translational control. Eur Respir J 54(2)
Clark KL, Li Y, Krauss MR, Kelley PW (2000) The asthma accession standard: a survival analysis of military recruits, 1995 to 1997. Mil Med 165(11):852–854
Liu H, Tan J, Liu J, Feng H, Pan D (2019) Altered mast cell activity in response to rhinovirus infection provides novel insight into asthma. J Asthma 18:1–9
Salomonsson M, Malinovschi A, Kalm-Stephens P, Dahlin JS, Janson C, Alving K, Hallgren J (2019) Circulating mast cell progenitors correlate with reduced lung function in allergic asthma. Clin Exp Allergy 49(6):874–882
Komi DEA, Bjermer L (2019) Mast Cell-mediated orchestration of the immune responses in human allergic asthma: current insights. Clin Rev Allergy Immunol 56(2):234–247
Petsky HL, Cates CJ, Kew KM, Chang AB (2018) Tailoring asthma treatment on eosinophilic markers (exhaled nitric oxide or sputum eosinophils): a systematic review and meta-analysis. Thorax 73(12):1110–1119
Cooper K, Frampton G, Harris P, Rose M, Chorozoglou M, Pickett K (2018) Reslizumab for Treating asthma with elevated blood eosinophils inadequately controlled by inhaled corticosteroids: an evidence review group perspective of a NICE single technology appraisal. pharmacoeconomics 36(5):545–553
Yoshikawa S, Oh-Hora M, Hashimoto R, Nagao T, Peters L, Egawa M, Ohta T, Miyake K, Adachi T, Kawano Y, Yamanishi Y, Karasuyama H (2019) Pivotal role of STIM2, but not STIM1, in IL-4 production by IL-3-stimulated murine basophils. Sci Signal 12(576)
Cirino M, Lagente V, Lefort J, Vargaftig BB (1986) A study with BN 52021 demonstrates the involvement of PAF-acether in IgE-dependent anaphylactic bronchoconstriction. Prostaglandins 32(1):121–126
Casale TB, Luskin AT, Busse W, Zeiger RS, Trzaskoma B, Yang M, Griffin NM, Chipps BE (2019) Omalizumab Effectiveness by biomarker status in patients with asthma: evidence from PROSPERO, a prospective real-world study. J Allergy Clin Immunol Pract 7(1):156–164 e1
Matucci A, Vultaggio A, Maggi E, Kasujee I (2018) Is IgE or eosinophils the key player in allergic asthma pathogenesis? Are we asking the right question? Respir Res 19(1):113
Rosenberg HF, Dyer KD, Foster PS (2013) Eosinophils: changing perspectives in health and disease. Nat Rev Immunol 13(1):9–22
Mesnil C, Raulier S, Paulissen G, Xiao X, Birrell MA, Pirottin D, Janss T, Starkl P, Ramery E, Henket M, Schleich FN, Radermecker M, Thielemans K, Gillet L, Thiry M, Belvisi MG, Louis R, Desmet C, Marichal T, Bureau F (2016) Lung-resident eosinophils represent a distinct regulatory eosinophil subset. J Clin Invest 126(9):3279–3295
Kopf M, Brombacher F, Hodgkin PD, Ramsay AJ, Milbourne EA, Dai WJ, Ovington KS, Behm CA, Kohler G, Young IG, Matthaei KI (1996) IL-5-deficient mice have a developmental defect in CD5+ B-1 cells and lack eosinophilia but have normal antibody and cytotoxic T cell responses. Immunity 4(1):15–24
Radermecker C, Sabatel C, Vanwinge C, Ruscitti C, Marechal P, Perin F, Schyns J, Rocks N, Toussaint M, Cataldo D, Johnston SL, Bureau F, Marichal T (2019) Locally instructed CXCR4(hi) neutrophils trigger environment-driven allergic asthma through the release of neutrophil extracellular traps. Nat Immunol
Ekstedt S, Stenberg H, Tufvesson E, Diamant Z, Bjermer L, Kumlien Georen S, Cardell LO (2019) The potential role of CD16(high) CD62L(dim) neutrophils in the allergic asthma. Allergy
Busse WW (2019) What are those neutrophils doing in severe asthma anyway? J Allergy Clin Immunol Pract 7(2):526–528
Grunwell JR, Stephenson ST, Tirouvanziam R, Brown LAS, Brown MR, Fitzpatrick AM (2019) Children with neutrophil-predominant severe asthma have proinflammatory neutrophils with enhanced survival and impaired clearance. J Allergy Clin Immunol Pract 7(2):516–525 e6
Radermecker C, Louis R, Bureau F, Marichal T (2018) Role of neutrophils in allergic asthma. Curr Opin Immunol 54:28–34
Saradna A, Do DC, Kumar S, Fu QL, Gao P (2018) Macrophage polarization and allergic asthma. Transl Res 191:1–14
Chung FT, Huang HY, Lo CY, Huang YC, Lin CW, He CC, He JR, Sheng TF, Wang CH (2019) Increased ratio of matrix metalloproteinase-9 (MMP-9)/Tissue inhibitor metalloproteinase-1 from alveolar macrophages in chronic asthma with a fast decline in FEV1 at 5-year follow-up. J Clin Med 8(9)
Tokunaga Y, Imaoka H, Kaku Y, Kawayama T, Hoshino T (2019) The significance of CD163-expressing macrophages in asthma. Ann Allergy Asthma Immunol 123(3):263–270
de Groot LES, van der Veen TA, Martinez FO, Hamann J, Lutter R, Melgert BN (2019) Oxidative stress and macrophages: driving forces behind exacerbations of asthma and chronic obstructive pulmonary disease? Am J Physiol Lung Cell Mol Physiol 316(2):L369–L384
Ubel C, Graser A, Koch S, Rieker RJ, Lehr HA, Muller M, Finotto S (2014) Role of Tyk-2 in Th9 and Th17 cells in allergic asthma. Sci Rep 4:5865
Chen Z, Wang L (2019) Ovalbumin induces natural killer cells to secrete Th2 cytokines IL5 and IL13 in a mouse model of asthma. Mol Med Rep 19(4):3210–3216
Leomicronn B (2017) T cells in allergic asthma: key players beyond the Th2 pathway. Curr Allergy Asthma Rep 17(7):43
Krabbendam L, Bal SM, Spits H, Golebski K (2018) New insights into the function, development, and plasticity of type 2 innate lymphoid cells. Immunol Rev 286(1):74–85
Nakamura Y, Ghaffar O, Olivenstein R, Taha RA, Soussi-Gounni A, Zhang DH, Ray A, Hamid Q (1999) Gene expression of the GATA-3 transcription factor is increased in atopic asthma. J Allergy Clin Immunol 103(2 Pt 1):215–222
Finotto S, De Sanctis GT, Lehr HA, Herz U, Buerke M, Schipp M, Bartsch B, Atreya R, Schmitt E, Galle PR, Renz H, Neurath MF (2001) Treatment of allergic airway inflammation and hyperresponsiveness by antisense-induced local blockade of GATA-3 expression. J Exp Med 193(11):1247–1260
Krug N, Hohlfeld JM, Kirsten AM, Kornmann O, Beeh KM, Kappeler D, Korn S, Ignatenko S, Timmer W, Rogon C, Zeitvogel J, Zhang N, Bille J, Homburg U, Turowska A, Bachert C, Werfel T, Buhl R, Renz J, Garn H, Renz H (2015) Allergen-induced asthmatic responses modified by a GATA3-specific DNAzyme. N Engl J Med 372(21):1987–1995
Ubel C, Sopel N, Graser A, Hildner K, Reinhardt C, Zimmermann T, Rieker RJ, Maier A, Neurath MF, Murphy KM, Finotto S (2014) The activating protein 1 transcription factor basic leucine zipper transcription factor, ATF-like (BATF), regulates lymphocyte- and mast cell-driven immune responses in the setting of allergic asthma. J Allergy Clin Immunol 133(1):198–206 e1-9
Jabeen R, Goswami R, Awe O, Kulkarni A, Nguyen ET, Attenasio A, Walsh D, Olson MR, Kim MH, Tepper RS, Sun J, Kim CH, Taparowsky EJ, Zhou B, Kaplan MH (2013) Th9 cell development requires a BATF-regulated transcriptional network. J Clin Invest 123(11):4641–4653
Jabeen R, Kaplan MH (2012) The symphony of the ninth: the development and function of Th9 cells. Curr Opin Immunol 24(3):303–307
Kaplan MH (2013) Th9 cells: differentiation and disease. Immunol Rev 252(1):104–115
Liao W, Spolski R, Li P, Du N, West EE, Ren M, Mitra S, Leonard WJ (2014) Opposing actions of IL-2 and IL-21 on Th9 differentiation correlate with their differential regulation of BCL6 expression. Proc Natl Acad Sci U S A 111(9):3508–3513
Licona-Limon P, Henao-Mejia J, Temann AU, Gagliani N, Licona-Limon I, Ishigame H, Hao L, Herbert DR, Flavell RA (2013) Th9 cells drive host immunity against gastrointestinal worm infection. Immunity 39(4):744–757
Neurath MF, Kaplan MH (2017) Th9 cells in immunity and immunopathological diseases. Semin Immunopathol 39(1):1–4
Levitt RC, McLane MP, MacDonald D, Ferrante V, Weiss C, Zhou T, Holroyd KJ, Nicolaides NC (1999) IL-9 pathway in asthma: new therapeutic targets for allergic inflammatory disorders. J Allergy Clin Immunol 103(5 Pt 2):S485–S491
Hoppenot D, Malakauskas K, Lavinskiene S, Sakalauskas R (2015) p-STAT6, PU.1, and NF-kappaB are involved in allergen-induced late-phase airway inflammation in asthma patients. BMC Pulm Med 15:122
Hoppenot D, Malakauskas K, Lavinskiene S, Bajoriuniene I, Kalinauskaite V, Sakalauskas R (2015) Peripheral blood Th9 cells and eosinophil apoptosis in asthma patients. Medicina 51(1):10–17
Finotto S (2018) B lymphocyte-induced maturation protein 1 (Blimp-1), a negative regulator of TH9 development, orchestrates the resolution of airway inflammation in patients with allergic asthma. J Allergy Clin Immunol
Erpenbeck VJ, Hohlfeld JM, Volkmann B, Hagenberg A, Geldmacher H, Braun A, Krug N (2003) Segmental allergen challenge in patients with atopic asthma leads to increased IL-9 expression in bronchoalveolar lavage fluid lymphocytes. J Allergy Clin Immunol 111(6):1319–1327
Abdelilah S, Latifa K, Esra N, Cameron L, Bouchaib L, Nicolaides N, Levitt R, Hamid Q (2001) Functional expression of IL-9 receptor by human neutrophils from asthmatic donors: role in IL-8 release. J Immunol 166(4):2768–2774
Bhathena PR, Comhair SA, Holroyd KJ, Erzurum SC (2000) Interleukin-9 receptor expression in asthmatic airways In vivo. Lung 178(3):149–160
Cheng G, Arima M, Honda K, Hirata H, Eda F, Yoshida N, Fukushima F, Ishii Y, Fukuda T (2002) Anti-interleukin-9 antibody treatment inhibits airway inflammation and hyperreactivity in mouse asthma model. Am J Respir Crit Care Med 166(3):409–416
Shimbara A, Christodoulopoulos P, Soussi-Gounni A, Olivenstein R, Nakamura Y, Levitt RC, Nicolaides NC, Holroyd KJ, Tsicopoulos A, Lafitte JJ, Wallaert B, Hamid QA (2000) IL-9 and its receptor in allergic and nonallergic lung disease: increased expression in asthma. J Allergy Clin Immunol 105(1 Pt 1):108–115
Toda M, Tulic MK, Levitt RC, Hamid Q (2002) A calcium-activated chloride channel (HCLCA1) is strongly related to IL-9 expression and mucus production in bronchial epithelium of patients with asthma. J Allergy Clin Immunol 109(2):246–250
Oh CK, Leigh R, McLaurin KK, Kim K, Hultquist M, Molfino NA (2013) A randomized, controlled trial to evaluate the effect of an anti-interleukin-9 monoclonal antibody in adults with uncontrolled asthma. Respir Res 14:93
Rubner FJ, Jackson DJ, Evans MD, Gangnon RE, Tisler CJ, Pappas TE, Gern JE, Lemanske RF Jr (2017) Early life rhinovirus wheezing, allergic sensitization, and asthma risk at adolescence. J Allergy Clin Immunol 139(2):501–507
Bergauer A, Sopel N, Kross B, Vuorinen T, Xepapadaki P, Weiss ST, Blau A, Sharma H, Kraus C, Springel R, Rauh M, Mittler S, Graser A, Zimmermann T, Melichar VO, Kiefer A, Kowalski ML, Sobanska A, Jartti T, Lukkarinen H, Papadopoulos NG, Finotto S (2017) Rhinovirus species/genotypes and interferon-lambda: subtypes, receptor and polymorphisms - missing pieces of the puzzle of childhood asthma? Eur Respir J 49(3)
Hansel TT, Tunstall T, Trujillo-Torralbo MB, Shamji B, del-Rosario A, Dhariwal J, Kirk PDW, Stumpf MPH, Koopmann J, Telcian A, Aniscenko J, Gogsadze L, Bakhsoliani E, Stanciu L, Bartlett N, Edwards M, Walton R, Mallia P, Hunt TM, Hunt TL, Hunt DG, Westwick J, Edwards M, Kon OM, Jackson DJ, Johnston SL (2017) A Comprehensive evaluation of nasal and bronchial cytokines and chemokines following experimental rhinovirus infection in allergic asthma: increased interferons (IFN-gamma and IFN-lambda) and type 2 inflammation (IL-5 and IL-13). EBioMedicine 19:128–138
Sykes A, Macintyre J, Edwards MR, Del Rosario A, Haas J, Gielen V, Kon OM, McHale M, Johnston SL (2014) Rhinovirus-induced interferon production is not deficient in well controlled asthma. Thorax 69(3):240–246
Bielor C, Sopel N, Maier A, Blau A, Sharma H, Vuorinen T, Kross B, Mittler S, Graser A, Mousset S, Melichar VO, Kiefer A, Zimmermann T, Springel R, Holzinger C, Trump S, Taka S, Papadopoulos NG, Weiss ST, Finotto S (2017) Role of TGF-beta in anti-rhinovirus immune responses in asthmatic patients. J Allergy Clin Immunol 140(1):283–286 e10
Usui T, Nishikomori R, Kitani A, Strober W (2003) GATA-3 suppresses Th1 development by downregulation of Stat4 and not through effects on IL-12Rbeta2 chain or T-bet. Immunity 18(3):415–428
Zhu J, Jankovic D, Oler AJ, Wei G, Sharma S, Hu G, Guo L, Yagi R, Yamane H, Punkosdy G, Feigenbaum L, Zhao K, Paul WE (2012) The transcription factor T-bet is induced by multiple pathways and prevents an endogenous Th2 cell program during Th1 cell responses. Immunity 37(4):660–673
Szabo SJ, Kim ST, Costa GL, Zhang X, Fathman CG, Glimcher LH (2000) A novel transcription factor, T-bet, directs Th1 lineage commitment. Cell 100(6):655–669
Neurath MF, Weigmann B, Finotto S, Glickman J, Nieuwenhuis E, Iijima H, Mizoguchi A, Mizoguchi E, Mudter J, Galle PR, Bhan A, Autschbach F, Sullivan BM, Szabo SJ, Glimcher LH, Blumberg RS (2002) The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J Exp Med 195(9):1129–1143
Finotto S, Neurath MF, Glickman JN, Qin S, Lehr HA, Green FH, Ackerman K, Haley K, Galle PR, Szabo SJ, Drazen JM, De Sanctis GT, Glimcher LH (2002) Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science 295(5553):336–338
Liu X, Li S, Jin J, Zhu T, Xu K, Liu C, Zeng Y, Mao R, Wang X, Chen Z (2019) Preventative tracheal administration of interleukin-27 attenuates allergic asthma by improving the lung Th1 microenvironment. J Cell Physiol 234(5):6642–6653
Joerger M, Finn SP, Cuffe S, Byrne AT, Gray SG (2016) The IL-17-Th1/Th17 pathway: an attractive target for lung cancer therapy? Expert Opin Ther Targets 20(11):1339–1356
Reppert S, Boross I, Koslowski M, Tureci O, Koch S, Lehr HA, Finotto S (2011) A role for T-bet-mediated tumour immune surveillance in anti-IL-17A treatment of lung cancer. Nat Commun 2:600
Xu L, Sun WJ, Jia AJ, Qiu LL, Xiao B, Mu L, Li JM, Zhang XF, Wei Y, Peng C, Zhang DS, Xiang XD (2018) MBD2 regulates differentiation and function of Th17 cells in neutrophils- dominant asthma via HIF-1alpha. J Inflamm 15:15
Vroman H, Bergen IM, van Hulst JAC, van Nimwegen M, van Uden D, Schuijs MJ, Pillai SY, van Loo G, Hammad H, Lambrecht BN, Hendriks RW, Kool M (2018) TNF-alpha-induced protein 3 levels in lung dendritic cells instruct TH2 or TH17 cell differentiation in eosinophilic or neutrophilic asthma. 141(5):1620–J Allergy Clin Immunol, 1633 e12
Wang L, Wan H, Tang W, Ni Y, Hou X, Pan L, Song Y, Shi G (2018) Critical roles of adenosine A2A receptor in regulating the balance of Treg/Th17 cells in allergic asthma. Clin Respir J 12(1):149–157
Guan Q, Yang B, Warrington RJ, Mink S, Kalicinsky C, Becker AB, Simons E, Peng Z (2019) Myeloid-derived suppressor cells: roles and relations with Th2, Th17, and Treg cells in asthma. Allergy
Ramakrishnan RK, Al Heialy S, Hamid Q (2019) Role of IL-17 in asthma pathogenesis and its implications for the clinic. Expert Rev Respir Med:1–12
Quan-San Z, Xiaohong X, Ying L, Zhaojia S (2019) Role of Th17-cell related cytokines in geriatric asthma. J Int Med Res 47(2):580–590
Zou XL, Chen ZG, Zhang TT, Feng DY, Li HT, Yang HL (2018) Th17/Treg homeostasis, but not Th1/Th2 homeostasis, is implicated in exacerbation of human bronchial asthma. Ther Clin Risk Manag 14:1627–1636
Massague J, Attisano L, Wrana JL (1994) The TGF-beta family and its composite receptors. Trends Cell Biol 4(5):172–178
Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, Rudensky AY (2009) CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 326(5955):986–991
DeVries A, Vercelli D (2018) Of pleiotropy and trajectories: Does the TGF-beta pathway link childhood asthma and chronic obstructive pulmonary disease? J Allergy Clin Immunol 141(6):1992–1996
Branchett WJ, Stolting H, Oliver RA, Walker SA, Puttur F, Gregory LG, Gabrysova L, Wilson MS, O'Garra A, Lloyd CM (2019) A T cell-myeloid IL-10 axis regulates pathogenic IFN-gamma-dependent immunity in a mouse model of type 2-low asthma. J Allergy Clin Immunol
Zonoobi E, Saeedfar K, Pourdowlat G, Masjedi MR, Behmanesh M (2018) The study of IL-10 and IL-17A genes expression in patients with different stages of asthma: a case-control study. Tanaffos 17(3):146–154
Paw M, Wnuk D, Kadziolka D, Sek A, Lasota S, Czyz J, Madeja Z, Michalik M (2018) Fenofibrate reduces the asthma-related fibroblast-to-myofibroblast transition by TGF-beta/Smad2/3 signaling attenuation and connexin 43-dependent phenotype destabilization. Int J Mol Sci 19(9)
Chen S, Han Y, Chen H, Wu J, Zhang M (2018) Bcl11b regulates IL-17 through the TGF-beta/Smad pathway in HDM-induced asthma. Allergy, Asthma Immunol Res 10(5):543–554
Haspeslagh E, Vanheerswynghels M, Deswarte K, Van Moorleghem J, Jacquet A, Lambrecht BN, Hammad H (2019) Prophylactic allergen immunotherapy with Der p 2 prevents murine asthma by regulating lung GM-CSF. J Allergy Clin Immunol 143(6):2307–2311 e5
Bohm L, Maxeiner J, Meyer-Martin H, Reuter S, Finotto S, Klein M, Schild H, Schmitt E, Bopp T, Taube C (2015) IL-10 and regulatory T cells cooperate in allergen-specific immunotherapy to ameliorate allergic asthma. J Immunol 194(3):887–897
Asamoah F, Kakourou A, Dhami S, Lau S, Agache I, Muraro A, Roberts G, Akdis C, Bonini M, Cavkaytar O, Flood B, Izuhara K, Jutel M, Kalayci O, Pfaar O, Sheikh A (2017) Allergen immunotherapy for allergic asthma: a systematic overview of systematic reviews. Clin Transl Allergy 7:25
Dhami S, Kakourou A, Asamoah F, Agache I, Lau S, Jutel M, Muraro A, Roberts G, Akdis CA, Bonini M, Cavkaytar O, Flood B, Gajdanowicz P, Izuhara K, Kalayci O, Mosges R, Palomares O, Pfaar O, Smolinska S, Sokolowska M, Asaria M, Netuveli G, Zaman H, Akhlaq A, Sheikh A (2017) Allergen immunotherapy for allergic asthma: a systematic review and meta-analysis. Allergy 72(12):1825–1848
MacDonald KM, Kavati A, Ortiz B, Alhossan A, Lee CS, Abraham I (2019) Short- and long-term real-world effectiveness of omalizumab in severe allergic asthma: systematic review of 42 studies published 2008-2018. Expert Rev Clin Immunol 15(5):553–569
Nishima S, Kozawa M, Milligan KL, Papadopoulos NG (2019) Omalizumab and unmet needs in severe asthma and allergic comorbidities in Japanese children. Asia Pac Allergy 9(1):e7
Deschildre A, Roussel J, Drumez E, Abou-Taam R, Rames C, Le Roux P, Pouessel G, Scalbert M, Bonnel C, Mitha S, Boileau S, Mordacq C, Thumerelle C, Labreuche J, Lejeune S, Marguet C (2019) Omalizumab discontinuation in children with severe allergic asthma: an observational real-life study. Allergy 74(5):999–1003
Fiocchi A, Artesani MC, Riccardi C, Mennini M, Pecora V, Fierro V, Calandrelli V, Dahdah L, Valluzzi RL (2019) Impact of omalizumab on food allergy in patients treated for asthma: a real-life study. J Allergy Clin Immunol Pract 7(6):1901–1909 e5
Weir E, Paton J (2019) Mepolizumab in adolescents with severe eosinophilic asthma not eligible for omalizumab: one center’s early clinical experience. J Asthma 22:1–4
Davila Gonzalez I, Moreno Benitez F, Quirce S (2019) Benralizumab: a new approach for the treatment of severe eosinophilic asthma. J Investig Allergol Clin Immunol 29(2):84–93
Chupp G, Lugogo NL, Kline JN, Ferguson GT, Hirsch I, Goldman M, Zangrilli JG, Trudo F (2019) Rapid onset of effect of benralizumab on morning peak expiratory flow in severe, uncontrolled asthma. Ann Allergy Asthma Immunol 122(5):478–485
Minami D, Kayatani H, Sato K, Fujiwara K, Shibayama T (2019) Effectiveness of benralizumab for allergic and eosinophilic predominant asthma following negative initial results with omalizumab. Respirol Case Rep 7(1):e00388
Busse WW, Bleecker ER, FitzGerald JM, Ferguson GT, Barker P, Sproule S, Olsson RF, Martin UJ, Goldman M, B.s. investigators (2019) Long-term safety and efficacy of benralizumab in patients with severe, uncontrolled asthma: 1-year results from the BORA phase 3 extension trial. Lancet Respir Med 7(1):46–59
Zayed Y, Kheiri B, Banifadel M, Hicks M, Aburahma A, Hamid K, Bachuwa G, Chandran A (2018) Dupilumab safety and efficacy in uncontrolled asthma: a systematic review and meta-analysis of randomized clinical trials. J Asthma 1:1–10
Corren J, Castro M, Chanez P, Fabbri L, Joish VN, Amin N, Graham NMH, Mastey V, Abbe A, Taniou C, Mahajan P, Teper A, Pirozzi G, Eckert L (2019) Dupilumab improves symptoms, quality of life, and productivity in uncontrolled persistent asthma. Ann Allergy Asthma Immunol 122(1):41–49 e2
Rabe KF, Nair P, Brusselle G, Maspero JF, Castro M, Sher L, Zhu H, Hamilton JD, Swanson BN, Khan A, Chao J, Staudinger H, Pirozzi G, Antoni C, Amin N, Ruddy M, Akinlade B, Graham NMH, Stahl N, Yancopoulos GD, Teper A (2018) Efficacy and safety of dupilumab in glucocorticoid-dependent severe asthma. N Engl J Med 378(26):2475–2485
Castro M, Corren J, Pavord ID, Maspero J, Wenzel S, Rabe KF, Busse WW, Ford L, Sher L, FitzGerald JM, Katelaris C, Tohda Y, Zhang B, Staudinger H, Pirozzi G, Amin N, Ruddy M, Akinlade B, Khan A, Chao J, Martincova R, Graham NMH, Hamilton JD, Swanson BN, Stahl N, Yancopoulos GD, Teper A (2018) Dupilumab efficacy and safety in moderate-to-severe uncontrolled asthma. N Engl J Med 378(26):2486–2496
Busse WW, Maspero JF, Rabe KF, Papi A, Wenzel SE, Ford LB, Pavord ID, Zhang B, Staudinger H, Pirozzi G, Amin N, Akinlade B, Eckert L, Chao J, Graham NMH, Teper A (2018) Liberty asthma QUEST: phase 3 randomized, double-blind, placebo-controlled, parallel-group study to evaluate Dupilumab efficacy/safety in patients with uncontrolled, moderate-to-severe asthma. Adv Ther 35(5):737–748
Weinstein SF, Katial R, Jayawardena S, Pirozzi G, Staudinger H, Eckert L, Joish VN, Amin N, Maroni J, Rowe P, Graham NMH, Teper A (2018) Efficacy and safety of dupilumab in perennial allergic rhinitis and comorbid asthma. J Allergy Clin Immunol 142(1):171–177 e1
Acknowledgments
The author thanks Julia Kölle, Adriana Geiger, Susanne Mittler, Eveldina Nendel, and Sonja Trump for their assistance and continuous commitment to our scientific research.
Funding
This work was also supported by a DFG grant FI817/6-1 awarded to Susetta Finotto and by the SFB1181.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The author declares that she has no conflict of interest.
Additional information
This article is a contribution to the special issue on Resolution of Inflammation in Chronic Diseases - Guest Editor: Markus Neurath
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Finotto, S. Resolution of allergic asthma. Semin Immunopathol 41, 665–674 (2019). https://doi.org/10.1007/s00281-019-00770-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00281-019-00770-3